Your new post is loading...
Your new post is loading...
University of Alabama at Birmingham Department of Medicine research results have identified a regulatory mechanism that controls the generation of germinal center-Tfh cells. The researchers believe their findings have important therapeutic implications.
Via Krishan Maggon
In the past 50 years, technological developments and intensive basic research have enabled substantial progress in our understanding of how the normal immune system works, how it participates in the pathophysiology of human diseases such as autoimmunity and immunodeficiencies, and how it can be pharmacologically influenced to combat diseases such as cancer, autoimmunity, inflammation, and infections. Arguably, the most detailed comprehension of the immune system concerns the activation and development of T cells and B cells, which collectively stipulate antigen‐specific immunity, by way of antigen‐specific helper or cytotoxic T cells and antigen‐specific antibody‐producing B cells. Beginning with the identification and gene cloning of the antigen receptors on T cells and B cells, research in this area has led to the characterization of multiple other receptors that diligently fine‐tune the activation and development of T cells and B cells, including co‐receptors, co‐stimulatory receptors, and co‐inhibitory receptors (Figure 1). Similarly, the intracellular signal transduction machineries coupling these receptors to cellular responses have been significantly elucidated. Mechanisms enabling diversification and specification of intracellular signals have been identified. More recent work, which has been facilitated by developments in microscopy technology and biophysical assays, has yielded an increasingly more detailed appreciation of the organization of signaling mechanisms at the molecular level, both in space and in time. A clearer picture of how alterations in these molecular pathways are linked to human diseases has ensued. In this issue of Immunological Reviews, leaders in the field review the literature and provide insights regarding various aspects of signal transduction and diversification in T cells and B cells. First, three reviews deal with the mechanisms by which engagement of the T‐cell antigen receptor (TCR) and other immune receptors leads to signal initiation. Wolgang Schamel (Freiburg, Germany) and collaborators review the main models of TCR conformational regulation. In essence, they converse on the current hypotheses regarding how ligand binding to the TCR changes the structure of the associated CD3‐ζ subunits. A unifying model is proposed. Etienne Gagnon (Montréal, Québec, Canada) and his colleague review the importance of electrostatic interactions in assembly, maintenance of inactive state, triggering and signaling by immune receptors, with a special emphasis on T cells. They discuss how the cytoplasmic domains of TCR subunits interact with charged lipids in the plasma membrane, and how these interactions are regulated in order to influence TCR signal initiation and strength. Furthermore, Enfui Hui (San Diego, CA, USA) and his collaborators review recent inroads into T‐cell signaling that have been provided by membrane reconstitution analyses. Notably, their own work has led to key new insights into how the inhibitory immune checkpoint receptor programmed death‐1 controls T‐cell activation. Next, two reviews focus on the mechanisms involved in amplification of antigen receptor‐driven signals. Claire Hivroz (Paris, France) and her colleagues review how TCR signaling is amplified and diversified in the endocytic compartment. They present data showing that the transmembrane adapter linker for activation of T cells (LAT) is shuttled between endocytic compartments and the immune synapse during T‐cell activation to provide signal amplification. Likewise, Wanli Liu (Beijing, China) and collaborators examine how the phospholipid phosphatidylinositol (PI) 4,5‐bisphosphate (PIP2) amplifies B‐cell antigen receptor (BCR)‐driven signals in B cells. Based on the studies discussed, the authors propose a “gasoline engine model” for amplification of B‐cell activation. They also review data showing the potential broad applicability of this model to other receptor signaling systems. In addition to antigen receptors, multiple other receptors and intracellular signaling molecules play essential roles in T‐cell and B‐cell activation and development. Four reviews cover specific aspects of this topic. Inducible costimulator (ICOS) is a receptor expressed on T cells that plays a critical role in co‐stimulation. Woong‐Kyung Suh (Montréal, Québec, Canada) and his team discuss in detail the roles, mechanisms of action, and signaling mechanisms of ICOS in immune cells, not only in follicular T‐helper (TFH) cells but also in other cell types. The authors also compare the functions and signaling mechanisms of ICOS with those of the related receptor CD28. Ion channels, including the calcium release‐activated channel (CRAC), have been broadly implicated in lymphocyte functions. Bebhinn Treanor (Toronto, Ontario, Canada) and colleagues review how these channels regulate B‐cell activation and development. They discuss not only calcium channels such as CRAC, but also chloride, potassium, and other types of ion channels. The Cbl proteins are ubiquitin ligases implicated in ubiquitination and degradation of several components of the antigen receptor signaling machinery in T cells and B cells. Hua Gu (Montréal, Québec, Canada) and his colleagues review accumulating data showing the broad range of molecular targets for the Cbl proteins in T cells and B cells, allowing the Cbl proteins to be gatekeepers of T‐cell activation, B‐cell development, and the germinal center reaction. Lastly, Jeroen Roose (San Francisco, CA, USA) and his collaborators review recent data showing how more downstream effectors of T‐cell activation, such as mammalian target of rapamycin and other kinases, are regulated to control T‐cell activation. The involvement of these kinases in mRNA translation, metabolic adaptation, and tonic signaling is also discussed. Last, three reviews deal with how alterations in components of the antigen receptor signaling machinery in T cells, B cells, or both have been broadly implicated in the pathophysiology of human diseases. Pamela Schwartzberg (Bethesda, MA, USA) and her team show that activating mutations in PI 3′ kinase in humans result in altered T‐cell and B‐cell functions, thereby causing a range of clinical manifestations including immunodeficiency and autoimmunity. In a related way, Sylvain Latour (Paris, France) and his collaborators examine how mutations in a broad array of components of the T‐cell signaling apparatus can cause immunodeficiencies in humans, especially upon infection by Epstein‐Barr virus. Some of these mutations affect proteins required for T‐cell cytotoxicity, while others involve polypeptides needed for T‐cell proliferation. Finally, Louis Staudt (Bethesda, MD, USA) and colleagues underscore how activating mutations in the BCR signaling machinery are implicated in human B‐cell malignancies. They discuss compelling clinical data showing that inhibitors of the mutated molecules can be used to treat effectively these diseases. Altogether, this monograph by leading scientists underscores the extensive progress that has been made through basic research in understanding how activation and development of T cells and B cells is controlled at the molecular level. It also clearly shows that alterations in the T‐cell or B‐cell signaling apparatus are implicated in human diseases. Furthermore, it indicates that dysregulated components of this machinery can be targeted to treat human pathologies. Nonetheless, while progress in this research area has been enormous over the past 50 years, it is clear that much more work in the future is needed to comprehend fully the processes involved, in order to use best the information provided for understanding and treating human diseases.
Summary Lymphoid organs guarantee productive immune cell interactions through the establishment of distinct microenvironmental niches that are built by fibroblastic reticular cells (FRC). These specialized immune‐interacting fibroblasts coordinate the migration and positioning of lymphoid and myeloid cells in lymphoid organs and provide essential survival and differentiation factors during homeostasis and immune activation. In this review, we will outline the current knowledge on FRC functions in secondary lymphoid organs such as lymph nodes, spleen and Peyer's patches and will discuss how FRCs contribute to the regulation of immune processes in fat‐associated lymphoid clusters. Moreover, recent evidence indicates that FRC critically impact immune regulatory processes, for example, through cytokine deprivation during immune activation or through fostering the induction of regulatory T cells. Finally, we highlight how different FRC subsets integrate innate immunological signals and molecular cues from immune cells to fulfill their function as nexus between innate and adaptive immune responses. 1 FIBROBLASTIC RETICULAR CELLS UNDERPIN THE STRUCTURE OF LYMPHOID ORGANS The activation of adaptive immune responses depends on the interaction of professional antigen‐presenting cells (APC) with T and B cells in specialized compartments of lymphoid organs. Secondary lymphoid organs (SLO) are strategically positioned at routes of pathogen invasion and thereby increase the likelihood of lymphocytes to encounter their cognate antigens at a particular location and during a certain time window. Lymph nodes are found at convergence points of afferent lymph vessels and surveil extracellular fluids from separate areas of peripheral tissue. Other classical SLO such as Peyer′s patches are located right below the area of the intestinal surface they surveil, while the splenic white pulp provides specialized niches for the development of immune responses against blood‐borne pathogens.1 Nonclassical SLO such as fat‐associated lymphoid clusters (FALC) serve as surveillance hubs of the body cavities by sampling the fluids that are secreted by mesothelial cells.2 Transient lymphoid aggregates that display T‐ and B‐cell zone segregation and that appear in inflamed tissues are referred to as tertiary lymphoid structures (TLS, also known as tertiary lymphoid organs).3 Fluid flow in lymphoid organs is sustained by endothelial stromal cells with blood endothelial cells facilitating the delivery of oxygen and nutrients via blood vessels and lymphatic endothelial cells granting drainage of extracellular liquids via lymphatic vessels. The structural integrity of all lymphoid organs is determined by fibroblastic stromal cells that build, for example, the capsule of lymph nodes or the spleen. In addition, specialized immune‐interacting fibroblasts, generally termed fibroblastic reticular cells (FRC), form the scaffold structures that underpin the distinct microenvironments required for efficient immune cell interactions (Figure 1). FRC crucially contribute to the functioning of the immune system through the secretion of homeostatic chemokines to coordinate the interaction between APC and lymphocytes and provide growth and survival factors that nurture both innate and adaptive lymphocytes.4, 5 Moreover, FRC are equipped with a large range of pattern recognition receptors6-8 that play a crucial role in the control innate immune reactions. The detection of pathogen‐associated molecular signatures by germline‐encoded receptors is a key determinant for the immune system to distinguish harmful molecules of foreign origin from inoccuous self‐molecules.9, 10 Sensing of microbial products by pattern recognition receptors expressed by APC such as dendritic cells and macrophages has been considered as a key step for the activation of the adaptive immune system.9, 11 However, recent studies have revealed that FRC in classical and nonclassical SLO actively shape adaptive immune responses through the integration of innate immunological signals.8, 12 In this review, we will highlight the ability of FRC to generate tissue‐specific microenvironmental niches that orchestrate complex immunological processes and discuss recent work that has revealed the role of FRC as an important nexus of innate and adaptive immune responses. 2 DIVERSITY OF FIBROBLASTIC RETICULAR CELLS IN LYMPHOID ORGANS The most commonly used distinction of endothelial and fibroblastic stromal cells of SLO relies on the expression of the endothelial marker CD31 (platelet endothelial cell adhesion molecule‐1, PECAM‐1) and the fibroblast marker podoplanin (PDPN).13 While PDPN expression captures the majority of FRC in lymph nodes, the expression of this marker in the spleen is restricted mainly to FRC in the T‐cell zone.14 Moreover, PDPN is a surface molecule that is expressed by fibroblasts in nonlymphoid tissues and in tumors15 rendering this marker unsuitable to specifically track lymphoid tissue FRC. In contrast, the promoter activity of the FRC signature genes Ccl19 and Cxcl13 is well‐suited to capture the complexity of the immune‐interacting fibroblasts in SLO.16, 17 Indeed, the Ccl19‐Cre model facilitates targeting of FRC in all relevant microenvironments in lymph nodes,16, 18, 19 in Peyer's patches12 and in the white pulp of the spleen.20 Likewise, the Cxcl13‐Cre/tdTomato transgene targets the majority of FRC in all SLO.17 The combination of such advanced transgenic mouse models with single‐cell RNA‐seq‐based analyses of lymph node7, 21 and splenic white pulp22 FRC will enable a series of novel studies to further explore the functional complexity of FRC in lymphoid organs. 2.1 The many shapes of FRC in classical secondary lymphoid organs While the differentiation trajectories of splenic white pulp FRC from perivascular progenitors have been delineated recently using Ccl19 promoter‐based cell fate mapping22 and lineage tracing,20 the origin of lymph node FRC has not yet been fully elucidated. Nevertheless, the aggregation of Ccl19‐Cre+ and Cxcl13‐Cre+ cells in the vicinity of blood vessels of the lymph node anlage16, 17 strongly suggests that lymph node FRC originate from myofibroblastic progenitors in the perivascular space. It appears that these precursor cells are able to generate the various FRC subsets that underpin the major compartments of the lymph node (Figure 1A). The lymph fluid from peripheral tissues that arrives to the lymph node via the afferent lymph vessels is drained through the subcapsular sinus which is lined by lymphatic endothelial cells and, beneath the subcapsular sinus floor, by several layers of marginal reticular cells (MRC).23 These cells can be distinguished from other FRC subsets in lymph nodes by the expression of mucosal vascular addressin cell adhesion molecule 1 (MAdCAM‐1), receptor activator of nuclear factor kappa‐Β ligand (RANKL, TNFSF11), and CXCL13.23 In the spleen, MRC support immune responsiveness by supporting the capture and delivery of antigens from the marginal zone to B‐cell follicles.24 The expression of RANKL by Ccl19‐Cre+ lymph node MRC has been shown to be critical for the integrity of lymphatic endothelial cells in the subcapsular sinus25 indicating that MRC actively participate in shaping the cellular environment of the lymph node. Likewise, splenic white pulp MRC impact the phenotype of their interacting immune cell partners. For example, Notch 2‐driven differentiation of marginal zone B cells and of Esam+ dendritic cells requires the expression of the Notch ligand Delta‐like (DL)1 in splenic Ccl19‐Cre+ cells,26 while RANK has been shown to maintain myeloid cell populations in the marginal zone to secure initiation of antiviral immune responses.27 As an extension from the MRC layer unfolds the conduit system through the lymph node parenchyma as a fibrillary network formed by FRC.28 Conduits both in lymph nodes and in the splenic white pulp are ensheathed by FRC and consist of a collagen‐rich core that is surrounded by a microfibrillar zone and a basement membrane.29, 30 The conduit system conveys low‐molecular‐weight substances such as chemokines and antigens from the lymph node subcapsular sinus through the T‐cell area toward high endothelial venules.30 A recent report revealed that IgM transiently gains access to the luminal side of the lymph node conduit system to facilitate rapid export of early‐response IgM antibodies out of the lymph node parenchyma.31 In sum, MRC of lymph nodes and the splenic white pulp shape the border region of lymphoid tissues through the dedicated interaction with immune cells and other stromal cells and by maintaining communication channels for rapid distribution of immunologically relevant information. It will be important to fully exploit existing and novel reticular cell targeting approaches (such as the CollagenVI‐Cre model32) to characterize the function of MRC in Peyer's patches. T‐cell zone reticular cells (TRC), ie, the FRC that co‐localize predominantly with T cells and dendritic cells, express PDPN and the signature chemokines CCL19 and CCL21.12-14, 22 It is important to reiterate that PDPN expression in lymph nodes can be found in almost all FRC subsets and that high PDPN expression is associated with maturation into fully immunocompetent FRC,16 while PDPN expression both in Peyer's patches and in the splenic white pulp is mainly found in TRC.12, 22 Both T cells and dendritic cells are in close contact with TRC and move along their projections within the T‐cell area.33 The mobility of dendritic cells is boosted by the ligation of the C‐type lectin‐like receptor 2 (CLEC‐2) with PDPN on TRC.34 Moreover, lymph node TRC are thought to function as the major source of the cytokine IL‐7 to promote T‐cell homeostasis.13 However, since IL‐7 can also be produced by lymphatic endothelial cells within lymph nodes35 and in the afferent lymphatics,36 the specific function of TRC‐derived IL‐7 for T‐cell sustenance and activation remains to be demonstrated. B‐cell follicles harbor distinct FRC subsets that produce CXCL13, the B cell‐attracting chemokine that binds to CXCR5.37 As mentioned above, CXCL13‐expressing MRC extend into the B cell follicle, while the most prominent reticular cell subset that underpins the B‐cell area has been named follicular dendritic cell (FDC).38 Due to their dendritic morphology, FDC have been assumed to be related to conventional dendritic cells, ie, to originate from bone marrow progenitors. Only recently, the progenitor cell of FDC has been revealed as perivascular myofibroblast that is characterized by the expression of platelet‐derived growth factor receptor β (Pdgfrb, CD140b).39 FDC in the B cell follicle can be identified by the expression the complement receptors CD21 and CD35 and can be found both in primary and secondary follicles.40 The main functions of FDC are the retention and presentation of particulate antigen on their surface to B cells using a broad range of FCγ‐receptors (ie, CD16, CD32)41 and the promotion of affinity maturation and somatic hypermutation of B cells in the germinal center reaction.42 FDC and probably other FRC subsets43 in the B‐cell follicle control the survival of B cells via B cell activation factor (BAFF) or transmembrane activator and CAML interactor (TACI).44 It has been suggested that the dark zone of the germinal center harbors a subset of CXCL12‐expressing reticular cells (CRC) that express PDPN and can be lineage‐traced using the Ccl19‐Cre and CD21‐Cre model.18 However, a unique phenotype of CRC could not be revealed using single‐cell RNA‐seq analysis7 indicating that the phenotypical distinction of FRC/FDC subsets underpinning the germinal center is still unclear. A main fraction of FRC express markers that are indicative for their perivascular location such as Itga7 (integrin α7), Pdgfrb (CD140b), and Acta2 (αSMA) in lymph nodes6, 7 and Ly6a (Sca‐1), Pdgfra (CD140a), and Vcam1 (CD106) in the spleen.22 It is likely that the perivascular reticular cell (PRC) fraction harbors the adult progenitor of all FRC subsets.22, 39 Other regions of the lymph node such as the deep cortical area appear to harbor a subset of FRC that is characterized by the expression of CCL21a, CXCL12, and LepR.19 This area of the lymph node is occupied by T cells, dendritic cells, and B cells suggesting that FRC acquire distinct phenotypical properties when they interact with multiple cell types. Indeed, FRC attain yet other properties when they co‐localize in medullary cords with macrophages, NK cells, and plasma cells.19 In this location, medullary reticular cells (medRC) express high levels of CXCL12, IL‐6, and BAFF and facilitate thereby the formation of dedicated niches for plasma cells.45 Single‐cell RNA‐seq analysis has confirmed the existence of at least two FRC subsets that localize in the medullary region indicating that medRC also promote the maintenance of NK cells in this region.7 Clearly, further studies are required to unveil the molecular properties and function of FRC subsets not only in the lymph node B‐cell niches but also in the different microenvironments of classical SLO. 2.2 Limited FRC heterogeneity in nonclassical SLO and TLS While the formation of classical SLO, ie, lymph nodes, splenic white pulp and Peyer's patches, is fully dependent on the presence of the lymphotoxin‐β receptor,46 the generation of nonclassical SLO (eg, FALC) or TLS (eg, inducible bronchus‐associated lymphoid tissue [BALT]) is largely independent of this pathway.2 For example, the formation of FALC requires the activation of stromal cells via the production of inflammatory cytokines such as the tumor necrosis factor (TNF), which are induced through the presence of microbiota in the intestine.47 Interestingly, the highly activated milieu of the intestinal lamina propria does not provide sufficient cytokine‐mediated stimulation to override the dependence of cryptopatch and isolated follicle formation on lymphotoxin‐β receptor signaling,48 indicating that the pathways employed in the generation of nonclassical SLOs are organ‐dependent. Likewise, TLS, which are locally inducible leukocytic aggregates that form in chronically inflamed nonlymphoid tissues,49 can form in different organs in a context‐dependent manner through triggering of inflammatory circuits involving IL‐17, IL‐6, IL‐1β, and/or IL‐22.50-53 In terms of structural organization and FRC content, both nonclassical SLO (Figure 1B) and TLS (Figure 1C) exhibit a reduced complexity when compared to the classical SLO. We will focus our review here on FALC and inducible BALT as examples of nonclassical SLOs and TLS, respectively, to highlight the few knowns and many unknowns of FRC biology in these compartments. FALC are located beneath the mesothelium and are surrounded by adipose tissues. A clear structural segregation of lymphocytes is not recognizable with a dense cluster of B cells being intermingled with CD4+ T cells and CD11b+ myeloid cells.54, 55 The main B cell population within FALC are B1 B cells that patrol body cavities and are the source of natural, low‐affinity immunoglobulin M (IgM) antibodies that bind to pathogenic bacteria.56 FALC also contain innate lymphoid cells (ILC), particularly type 2 ILCs and NKT cells.47, 55 The production of CCL19 and CCL21 by FALC FRC most likely mediates the attraction and retention of naive T cells.54 PDPN‐expressing FRC that underpin FALC structures are highlighted by the Ccl19‐Cre transgene, express PDPN and occupy perivascular niches with a surface marker profile that resembles splenic PRC (ie, PDGFRα+VCAM‐1+ICAM‐1+).8 Although these cells do not display the general phenotype of MRC or FDC, FALC FRC have been shown to produce the B cell‐attractant CXCL13.57 Hence, it appears that the somewhat random mixture of T and B cells in FALC is due to a high versatility of FALC FRC which permits attraction and interaction with B cells, T cells, and myeloid cells. Clearly, FALC FRC—and probably as well the FRC underpinning intestinal isolated lymphoid follicles—can steer both innate and adaptive immune responses without forming distinct microenvironments such as germinal centers. The formation of TLS is frequently associated with chronic inflammatory and autoimmune diseases.3, 58 Importantly, in the context of cancer, the presence of TLS correlates with improved survival in a growing list of human cancers including breast cancer,59 lung cancer,60 oral squamous cell61 and Merkel cell carcinomas,62 and melanoma.63 Hence, it is tempting to speculate that TLS serve as inducible and transient outposts of the immune system to locally cope with ongoing immunological threats. Moreover, it appears that the coordination of immune cell interaction within these structures relies on organizational principles that are comparable to those in the classical SLO (Figure 1C). During tumor formation, TLS undergo a maturation process that has been suggested to start with the segregation of T‐cell and B‐cell areas in the perivascular space and progresses by the appearance of germinal centers.64, 65 The subsequent development of germinal centers in tumor TLS is accompanied by the appearance of CXCL13‐producing, CD21+ FDC networks both in human colorectal cancer65 and in squamous cell carcinoma of the lung.64 The presence of chemokine‐secreting FRC that underpin TLS has been demonstrated in a variety of models of chronic organ inflammation.52, 53, 66 The formation of inducible BALT in the lung has revealed that the activation of the innate immune system via lipopolysaccharide instillation can drive the formation of local TLS in an IL‐17‐dependent fashion,50 while this cytokine is not necessary to induce BALT formation following intranasal infection with a propagation‐deficient virus.67 Nevertheless, viral infection appears to be sufficient to induce highly organized BALT structures that contain B‐cell follicles underpinned by a network of CXCL13‐expressing FDC as well as CXCL12‐producing, yet undefined, reticular stromal cells.66 Furthermore, the bacterial infection with Pseudomonas aeruginosa triggers BALT formation in a TLR‐dependent manner leading to the emergence of CXCL12+ reticular cells that is dependent on γδ T cell‐derived IL‐17, while FDC fail to develop under these conditions.66 Overall, the emerging view is that remodeling and maturation of immune‐stimulating FRC is one of the initiating events in the establishment of an immune‐competent niche capable of recruiting and retaining disease‐relevant lymphocytes in TLS.3 Hence, targeted modulation of FRC differentiation processes within TLS may lead to treatment modalities that either attenuate TLS formation during chronic inflammatory diseases or foster the development of such immune‐activating structures in malignant diseases. 3 IMMUNE CELL‐FRC INTERACTIONS The main function of secondary lymphoid organs is to preempt68 and to deal with1 the encounter of microbial agents and tumor cells. Pathogens and other antigenic material arrive at the antigen‐sampling zone of SLO, eg, the subcapsular sinus region of lymph nodes, where dedicated macrophage/dendritic cell populations take up and transfer antigen to B cells.69, 70 Consequently, disruption of the subcapsular sinus structure results in defective immune responses during secondary infection in mice.71 The cellular infrastructure of the murine splenic marginal zone functions in the same manner and efficiently retains infectious agents.72 Interestingly, it appears that the function of antigen capture and innate immunological sensing in the marginal zone of murine spleens is assigned to CD169‐positive macrophages and/or dendritic cells,69, 72 while in the human spleen MAdCAM1‐positive MRC operate as coordinators of immune cell interaction and drivers of subsequent immune activation.24 Antigens are further dispersed in the lymph node through the lymphatic sinuses that pervade the lymph node parenchyma, and are taken up by distinct dendritic cell subsets for the delivery to CD8+ or CD4+ T cells.73 The interaction of dendritic cells and T cells depends on the infrastructure provided by TRC74 and is regulated by TRC‐derived factors such as CCL2175 or lysophosphatidic acid.76 The efficient interaction of T and B cells at the T‐B border during viral infection depends on the presence of BAFF‐producing FRC.44 Other FRC subsets contribute during the subsequent steps of B‐cell activation in lymph nodes including antigen presentation in the germinal center,42 regulating B‐cell migration during the germinal center reaction,18, 42 and establishing plasma cell competent microenvironments in the medulla.45 FRC can perform these multiple functions because they are able to integrate a variety of signals through sensing of innate immunological stimuli and the differentiation of cellular signals in their immediate environment (Figure 2A). In the following sections, we will summarize how FRC detect and process pathogen‐derived innate immunological signals and illustrate the molecular pathways employed by FRC to regulate adaptive immune responses in lymph nodes, Peyer's patches and FALC. 3.1 Innate immunological sensing by FRC FRC can directly recognize pathogens and their immune‐activating signals using various pattern recognition receptors and particular sets of immune‐activating molecules.6, 7, 77 The activation of in vitro cultivated FRC from SLO12 or FALC8 with various TLR ligands including lipopolysaccharide, poly(I:C), and zymosan leads to upregulation of adhesion molecules ICAM‐1 and VCAM‐1 and the secretion of inflammatory mediators such as CCL2, IL‐6, and TNF. In vivo, lymph node FRC react rapidly to systemic application of lipopolysaccharide with the activation of antigen presentation and type 1 interferon (IFN) pathways and alterations in the generation of extracellular matrix proteins including matrix metalloproteinase‐9, periostin, collagen type VI, and laminin α2.6 Direct ligation of TLR4 on FDC by lipopolysaccharide leads to increased expression of adhesion molecules and promotes the production of antigen‐specific antibodies when FDC are co‐cultured with B cells in vitro.78 Moreover, the activation of FDC by oxidized phospholipids, which function as endogenous TLR4 ligands, can foster the germinal center reaction by promoting higher rates of class‐switch recombination and somatic hypermutation in B cells.79 The formyl peptide receptor 2 that binds microbial products derived from Escherichia coli or Listeria, interacts with the endogenous ligand LL‐37 to enhance CXCL13 and BAFF production by FDC and thereby promotes B‐cell proliferation.80 Viruses can directly infect FRC as demonstrated for the lymphocytic choriomeningitis virus in mice14, 81 and human viruses including Chikungunya virus82 or Ebola virus.83 Complex immune cell interactions are triggered when intracellular viral RNA is recognized by FRC via TLR712 (Figure 2B). The transcriptomic analysis of lymph node FRCs after subcutaneous infection with herpes simplex virus‐1 (HSV‐1) revealed a pronounced activation of type‐I IFN pathway in FRC.77 Virus‐induced inflammation results in FRC proliferation and the induction of a substantial remodeling of the FRC landscape.35, 77 However, excessive activation of TLR7 ligands, eg, through internalization of ribonucleotide proteins complexes via CD21, can result in type 1 IFN production by FDC which supports the long‐lasting maintenance of the germinal center response and sustained production of antinuclear antibodies with perpetuation of a lupus‐like disease in mice.84 Hence, FRC activation through innate immunological pathways requires dedicated control mechanisms to avoid immunopathological sequela. Innate immunological recognition circuits are integrated intracellularly via particular molecular switches such as the myeloid differentiation primary response 88 (MYD88) protein85 and can be amplified by cellular receptors such as the type‐I IFN receptor (IFNAR).86 During infection with the murine cytomegalovirus, blockade of the type‐I IFN pathway leads to a change in viral tropism with a shift from subcapsular macrophages to MRC as the main target cells. The elevated infection rate of MRC leads to the activation and recruitment of NK cells, which efficiently reduce the viral burden in the subcapsular sinus but consequently destroy the reticular cell network of lymph nodes leading to systemic distribution of the virus.87 Overall, it appears that IFNAR signaling in the stromal cell compartment is important to contain viral replication in a broad range of experimental models. However, whether and to which extent IFNAR signaling in FRC directly contributes to the control of a viral infection has not been determined yet. Further FRC innate activation signals can be derived from immune cells that populate their particular microenvironmental niches. For example, in murine FALC, both FRC and hematopoietic cells attracted by FRC can serve as source for TNF8 (Figure 2C). Likewise, human tonsillar FRC respond in vitro to TNF exposure with increased expression of adhesion molecules and enhanced production of inflammatory cytokines.88 Further consequences of innate immunological sensing by FRC include the production of T cell‐activating factors such as IL‐33, which is produced by splenic PDPN+ FRC during vaccination with a recombinant viral vector.89 Single‐cell RNA‐seq analysis has revealed a population of FRC in naive lymph nodes that express high levels of Cxcl9.7 It appears that CXCL9 is mainly provided by stromal cells while CXCL10 is mainly express by murine myeloid cells DC following immunization of mice with dendritic cells that are pulsed with ovalbumin protein and activated with lipopolysaccharide and poly(I:C).90 In sum, FRC actively participate in the earliest phases of developing immune responses through their function as recipients of innate immunological signals and as coordinators of subsequent immune reactions through autocrine and paracrine signal amplification. 3.2 FRC control adaptive immune responses in lymph nodes and Peyer's patches Molecules of the TNF receptor superfamily are not only crucial for the formation of lymphoid organs2, 46, 68, 91 but also profoundly impact the maturation of myofibroblastic progenitors into fully immunocompetent FRC. For example, genetic ablation of lymphotoxin‐β receptor expression on Ccl19‐Cre+ FRC leads to defective FRC maturation with reduced expression of key molecules such as Ccl19, Ccl21, and Il7, that precipitates high susceptibility to viral infection due to impaired activation of T cells.16 While the lymphotoxin‐β receptor appears to affect the maturation of all FRC subsets in lymph nodes, other molecules from the TNF receptor superfamily appear to be required for subset specification. For example, FDC differentiation depends on TNF produced by B cells,92 while CD30 contributes to proper B‐ and T‐cell zone segregation that is associated with reduced PDPN expression on yet undefined FRC subsets.93 The intricate regulatory circuits of FRC‐immune cell interaction that found the basis of protective immune responses is highlighted in a recent study on intestinal Listeria monocytogenes infection in mice.94 Listeria infection induces intestinal epithelial cell proliferation and depletion of goblet cells, while CX3CR1+ myeloid cells in Peyer's patches produce IL‐23 and thereby activate IL‐17‐secreting γδ T cells. The resulting IL‐11 production by PDPN‐expressing cells in Peyer's patches facilitates the activation of enterocytes and limits intestinal villus invasion by Listeria.94 However, such protective FRC‐immune cell interactions may precipitate immunopathological consequences such as a decreased thickness of the mucus barrier that eventually fosters intestinal inflammation.94 Thus, FRC can function both as activating and regulating cellular entities during immune responses. The ability of FRC to negatively impact T‐cell responses has been first noted by the Turley group who demonstrated that FRC in lymph nodes can present peripheral tissue antigens to T cells and thereby attenuate self‐reactivity.95 It has been proposed that the generation of nitric oxide by lymph nodes FRC regulates the expansion and activity of T cells.96, 97 However, since nitric oxide can be produced by other lymph node stromal cells such as lymphatic endothelial cells,96 it remains to be determined, for example, through FRC‐specific ablation of the inducible nitric oxide synthase gene, to which extent FRC regulate T‐cell activity through this mechanism. It is also possible that FRC regulate global immune responsiveness by impacting regulatory T‐cell differentiation. It is generally assumed that dendritic cells are the main cell population that control the differentiation pathway of CD4+ T cells toward the FoxP3+ regulatory T‐cell phenotype.98, 99 However, a recent study suggests that FRC in mesenteric lymph nodes can modulate resident dendritic cells via a bone morphogenic protein‐2‐dependent pathway to foster the induction of regulatory T cells.21 Further studies are warranted to elaborate such direct and indirect pathways of FRC‐dependent immune regulation. Innate lymphoid cells (ILC) regulate immune responsiveness by bridging innate and adaptive immunity. In contrast to T and B cells, ILC lack rearranged antigen receptors and their development and activation is therefore mainly steered via soluble factors and their receptors. Since ILC are particularly abundant at mucosal sites, they are considered as the main cell population that maintains tissue integrity and homeostasis via innate immune mechanisms.100 ILC accumulation at mucosal surfaces relies on the provision of survival factors such as IL‐7 and IL‐15 that can be produced by FRC13, 101 However, ILC also reside in SLO where they are involved in the regulation of adaptive immunity.102, 103 Recently, it has been shown that group 1 ILC are localized in the T cell zones of Peyer's patches and that Ccl19‐Cre+ FRC generate an essential niche for these cells through the provision of IL‐15.12 However, IL‐15 production by PDPN+ FRC in Peyer's patches is rapidly abrogated under excessive inflammatory conditions such as infection with a cytopathic virus. Importantly, the swift cessation of IL‐15 production by FRC in Peyer's patches and mesenteric lymph nodes is dependent on MyD88 signaling which prevents an overshooting activation of NK1.1+ ILC and immunopathological overstimulation of IFN‐γ‐producing Th1 cells (Figure 2B). As a consequence, unrestrained Peyer's patch FRC lacking MyD88 expression permit the rapid clearance of a cytopathic viral infection through boosted ILC1 and Th1 responses which is accompanied by impaired intestinal integrity, bacterial dysbiosis, and chronical intestinal inflammation.12 This study shows that FRC in lymph nodes and Peyer's patches can act as immune rheostats through the regulation of group 1 ILC activity. It will be important to further elaborate the mechanisms that grant FRC in lymphoid organs control over innate and adaptive immune responses. 3.3 FRC‐dependent immune responses in FALC In case of a breach of pathogenic microorganisms into one of the body cavities, protective immunity needs to be mounted swiftly to prevent harm to the internal organs. The adipose tissue underlying the mesothelial surface of the pericardial, pleural and peritoneal cavities harbors variable numbers of FALC.104 In the omentum, a mesothelium‐covered tissue flap that connects stomach, pancreas, colon, and spleen, FALC have been shown to collect fluids and particles from the peritoneal cavity.105 The uptake of inflammation‐inducing substances from the peritoneal cavity such as lipopolysaccharide106 or the lipid‐based antigen zymosan47 induces an increase of omental FALC size and numbers. It has been shown that opsonization of bacterial antigens by natural, low‐affinity IgM antibodies generated by peritoneal B1 B cells can promote the uptake and elimination of bacteria by myeloid cells.107 Moreover, the presence of viral particles in the peritoneal cavity increases the cellularity and size of FALC with concomitant attraction of macrophages from the peritoneal cavity to these structures.108 Other immune cells in the peritoneal cavity such as B‐2 B cells109 or neutrophils110 reach FALC via the blood vasculature. Although FALC lack the compartmentalization observed in classical SLO, their structural foundations support the rapid generation of both T‐ and B‐cell immune responses. For example, intraperitoneal application of antigens such as ovalbumin leads to the initial activation of antigen‐specific CD4+ and CD8+ T cells in omental FALC.54 Similarly, T‐dependent and T‐independent B‐cell responses are generated first in FALC following intraperitoneal antigen application.47 The role of FRC in the initiation of adaptive immune responses in FALC has only recently been clarified. It appears that CXCL13‐expressing FRC in FALC not only support the attraction and retention of B cells,54, 57 but that Ccl19‐Cre+ FALC FRC can directly sense the presence of microbial products via TLR4 and initiate a MyD88‐dependent immune‐amplifying cascade8 (Figure 2C). Following exposure to TLR2 and TLR4 ligands FALC FRC secrete inflammatory cytokines and chemokines including CCL2 to attract CCR2+ inflammatory monocytes from the circulation.8 Inflammatory monocytes establish a crucial crosstalk with FALC FRC that leads to the rapid growth and remodeling of FALC. TNF functions as the main factor that regulates the reciprocal communication between FRC and the myeloid cells and eventually facilitates the generation of humoral immunity within FALC8, 47 (Figure 2C). Interestingly, although FALC lack FDC, the microenvironmental remodeling provided by FRC is sufficient to promote germinal center‐like B‐cell responses with class‐switch recombination and a discrete somatic hypermutation.47, 54 Currently, it remains unknown whether CXCL13+ and CCL19+ FRC represent two distinct cell types or whether a variable expression of the two chemokines represents different functional states. It will be possible to clarify this question by using appropriate cell fate mapping models to track the lineage commitments of FRC subsets within nonclassical SLO. Beyond their prominent function in the initiation and coordination of innate and adaptive immune responses in FALC, FRC present in visceral adipose tissues may also contribute to the maintenance of immune homeostasis and the regulation of the immune‐suppressive environment of adipose tissues under steady‐state conditions. Adipose tissues, including the omental fat favor the accumulation of IL‐10‐producing B cells111 and regulatory T cells.112 Regulatory T cells in adipose tissues are characterized by high expression of the IL‐33 receptor ST2,112 with IL‐33 being one of the main tissue factors that impacts the development and maintenance of regulatory T cells in this compartment.113 Since IL‐33 is mainly produced by FRC‐like cells in FALC,114 it is tempting to speculate that this circuit not only controls B1 B‐cell activation and local IgM production during lung infection and inflammation,114 but that FALC FRC equilibrate immune‐activating and ‐suppressive circuits in these tissues. The regulation of physiological functions by FRC‐derived IL‐33 in the adipose tissue may even extend to other body functions such a thermogenesis.115 Further studies will be required to better understand the molecular processes underlying FRC‐dependent immune activation in FALC. Moreover, it will be important to dissect the relation of FALC FRC to other PDPN expression fibroblasts in visceral adipose tissues and to elaborate the mechanisms employed by FRC that contribute to the maintenance of tissue homeostasis. 4 CONCLUDING REMARKS The translation of innate immune signals into activating or regulating processes that steer adaptive immune responses has been regarded as one of the main roles of professional APC such as dendritic cells. FRC as dedicated immune‐interacting fibroblasts have now entered the stage to be recognized as cells that crucially contribute to the decision‐making within the immune system. The many functions of FRC are accomplished through the generation of specific microenvironmental niches for various types of immune cells. Distinct subsets of FRC form these niches to support immune cell migration, survival, and differentiation. Interestingly, it seems that particular genetic programs are imprinted in the immune‐interacting fibroblasts depending on their anatomical location. The main differentiation switches of FRC differentiation have been identified with the lymphotoxin‐β receptor representing a necessary “signal 1” for FRC maturation in classical SLO.16 However, further research needs to dissect the intercellular communication pathways between FRCs and the various immune cells that impact the differentiation trajectories of FRC in lymphoid and nonlymphoid organs. Indeed, it appears that the intestinal lamina propria of patients suffering from inflammatory bowel disease harbors at least one mesenchymal stromal cell population that closely resembles lymphoid organ FRC and is highlighted by the expression of IL‐33 and Lysyl oxidases.116 Thus, understanding the mechanisms that govern FRC differentiation in SLO and TLS will, for example, open novel avenues to target drugable FRC differentiation pathways in TLS that form during human chronic inflammatory diseases such as rheumatoid arthritis117, 118 or Sjören's syndrome.119 A major challenge that should to be addressed in the future is the lineage relationship of FRC with other mesenchymal cell types such as adipocytes, chondrocytes, or regular tissue fibroblasts. The definition of mesenchymal cell types needs to be coupled to their development origin and their function within a tissue. Hence, a combination of single‐cell transcriptome analysis and faithful in vivo lineage tracing needs to be employed120 to obtain consistent definitions of the cell types that are currently covered by the broad terms “stromal cells” or “mesenchymal cells”. ACKNOWLEDGEMENTS We thank Dr. Natalia Pikor for critical reading of the manuscript. This study received financial support from the Swiss National Science Foundation (grants 166500 and 177208 to BL). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. CONFLICT OF INTEREST The authors declare no conflict of interest. REFERENCES
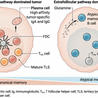
Key events in T cell-dependent antibody responses, including affinity maturation, are dependent on the B cell’s presentation of antigen to helper T cells at critical check points in germinal center formation in secondary lymphoid organs.
Germinal center (GC) B cells are essential to generating protective antibody responses and are selected through a process of affinity maturation. Kwak et al . now define intrinsic properties of human GC B cells that are critical to antigen affinity discrimination. They identified BCR-containing actin-rich pod-like structures that facilitated formation of highly stable synapses and antigen internalization but only when GC B cells engaged high-affinity antigens. These findings reveal the importance of these structures in setting thresholds for affinity selection and driving GC B cell responses.
Protective antibody responses to vaccination or infection depend on affinity maturation, a process by which high-affinity germinal center (GC) B cells are selected on the basis of their ability to bind, gather, and present antigen to T follicular helper (Tfh) cells. Here, we show that human GC B cells have intrinsically higher-affinity thresholds for both B cell antigen receptor (BCR) signaling and antigen gathering as compared with naïve B cells and that these functions are mediated by distinct cellular structures and pathways that ultimately lead to antigen affinity– and Tfh cell–dependent differentiation to plasma cells. GC B cells bound antigen through highly dynamic, actin- and ezrin-rich pod-like structures that concentrated BCRs. The behavior of these structures was dictated by the intrinsic antigen affinity thresholds of GC B cells. Low-affinity antigens triggered continuous engagement and disengagement of membrane-associated antigens, whereas high-affinity antigens induced stable synapse formation. The pod-like structures also mediated affinity-dependent antigen internalization by unconventional pathways distinct from those of naïve B cells. Thus, intrinsic properties of human GC B cells set thresholds for affinity selection.
PD-1 functions on T follicular helper cells to dictate localization within lymph node germinal centers. Program cell death protein 1 (PD-1) is an inhibitory B7 family member; it has been extensively studied for its role in inhibiting CD8+ T cell function within tumors or during chronic infections. This work has led to successful clinical use of checkpoint inhibitors to rejuvenate the antitumor T cell response. PD-1 is also a defining marker of CD4+ T follicular helper (TFH) cells, which are responsible for guiding B cell antibody production. What role it plays on TFH cell function is not well understood. Consistent with its inhibitory function, PD-1 does in fact constrain TFH cell development. However, recent work from Shi et al. demonstrates that PD-1 is also required to fine-tune the TFH-B cell humoral response by regulating TFH cell positioning within lymph nodes (LNs). As CD4+ T cells approach the follicle, they are rebuffed by bystander B cells expressing a ligand for PD-1, which dampens phosphoinositide-3 kinase (PI3K) signaling downstream of the chemokine receptor CXCR5 and thus slows T cell motility. As inducible T-cell costimulator (ICOS) enhances PI3K signals, theoretically only T cells with the highest level of ICOS can overcome this and make it through the inhibitory ring of B cells to the germinal center. PD-1 expression also blocks developing TFH cell distraction from chemokines expressed outside of the follicle by negatively regulating CXCR3 expression. All together then, PD-1 expression by recently activated CD4+ T cells helps to enforce a TFH cell concentration within the antigen-reactive B cell germinal center. An unresolved immunologic mystery is how naïve T cells—after early antigenic stimulation—“choose” a particular differentiation fate. In particular, T helper type 2 (TH2) and TFH cells require similar costimulatory signals from antigen presenting cells, express higher levels of CXCR5 than other T effector fates, and are concentrated in the same region of the LN during activation—the T-B border—where they are activated by type 2 conventional dendritic cells (cDC2s). Based on this recent work, differential expression of ICOS might be the tipping point toward TFH rather than TH2 differentiation. By overcoming PD-1 inhibition, strong ICOS signaling allows passage into the germinal center where the final stage of TFH differentiation occurs. Highlighted Article -
- J. Shi,
- S. Hou,
- Q. Fang,
- X. Liu,
- X. Liu,
- H. Qi
, PD-1 controls follicular T helper cell positioning and function.Immunity 49, 264– 274 ( 2018). Google Scholar - Copyright © 2018, American Association for the Advancement of Science
Via Krishan Maggon
Summary
Follicular T helper (Tfh) cells highly express the programmed cell death-1 (PD-1) molecule. Whereas inhibition of T cell receptor (TCR) signaling and CD28 co-stimulation is thought to be the primary mode of PD-1 functions, whether and how PD-1 regulates Tfh cell development and function is unclear. Here we showed that, when engaged by the ensemble of bystander B cells constitutively expressing PD-1 ligand 1 (PD-L1), PD-1 inhibited T cell recruitment into the follicle. This inhibition involved suppression of PI3K activities downstream of the follicle-guidance receptor CXCR5, was independent of co-signaling with the TCR, and necessitated ICOS signaling to overcome. PD-1 further restricted CXCR3 upregulation on Tfh cells, serving to concentrate these cells toward the germinal center territory, where PD-L1-PD-1 interactions between individual Tfh and B cells optimized B cell competition and affinity maturation. Therefore, operating in both costimulation-independent and -dependent manners, PD-1 controls tissue positioning and function of Tfh cells.
Via Krishan Maggon
The production of high-affinity antibody is essential for pathogen clearance. Antibody affinity is increased through germinal center (GC) affinity maturation, which relies on BCR somatic hypermutation (SHM) followed by antigen-based selection. GC B cell proliferation is essentially involved in these processes; it provides enough templates for SHM and also serves as a critical mechanism of positive selection. In this study, we show that expression of epigenetic regulator ubiquitin-like with PHD and RING finger domains 1 (Uhrf1) was markedly up-regulated by c-Myc–AP4 in GC B cells, and it was required for GC response. Uhrf1 regulates cell proliferation–associated genes including cdkn1a , slfn1 , and slfn2 by DNA methylation, and its deficiency inhibited the GC B cell cycle at G1-S phase. Subsequently, GC B cell SHM and affinity maturation were impaired, and Uhrf1 GC B knockout mice were unable to control chronic virus infection. Collectively, our data suggest that Uhrf1 regulates GC B cell proliferation and affinity maturation, and its expression in GC B cells is required for virus clearance.
Activated B cells can initially differentiate into three functionally distinct fates—early plasmablasts (PBs), germinal center (GC) B cells, or early memory B cells—by mechanisms that remain poorly understood. Here, we identify atypical chemokine receptor 4 (ACKR4), a decoy receptor that binds and degrades CCR7 ligands CCL19/CCL21, as a regulator of early activated B cell differentiation. By restricting initial access to splenic interfollicular zones (IFZs), ACKR4 limits the early proliferation of activated B cells, reducing the numbers available for subsequent differentiation. Consequently, ACKR4 deficiency enhanced early PB and GC B cell responses in a CCL19/CCL21-dependent and B cell–intrinsic manner. Conversely, aberrant localization of ACKR4-deficient activated B cells to the IFZ was associated with their preferential commitment to the early PB linage. Our results reveal a regulatory mechanism of B cell trafficking via an atypical chemokine receptor that shapes activated B cell fate.
Publication date: Available online 14 December 2017
Source:Cell
Author(s): Mauro Gaya, Patricia Barral, Marianne Burbage, Shweta Aggarwal, Beatriz Montaner, Andrew Warren Navia, Malika Aid, Carlson Tsui, Paula Maldonado, Usha Nair, Khader Ghneim, Padraic G. Fallon, Rafick-Pierre Sekaly, Dan H. Barouch, Alex K. Shalek, Andreas Bruckbauer, Jessica Strid, Facundo D. Batista
B cells constitute an essential line of defense from pathogenic infections through the generation of class-switched antibody-secreting cells (ASCs) in germinal centers. Although this process is known to be regulated by follicular helper T (TfH) cells, the mechanism by which B cells initially seed germinal center reactions remains elusive. We found that NKT cells, a population of innate-like T lymphocytes, are critical for the induction of B cell immunity upon viral infection. The positioning of NKT cells at the interfollicular areas of lymph nodes facilitates both their direct priming by resident macrophages and the localized delivery of innate signals to antigen-experienced B cells. Indeed, NKT cells secrete an early wave of IL-4 and constitute up to 70% of the total IL-4-producing cells during the initial stages of infection. Importantly, the requirement of this innate immunity arm appears to be evolutionarily conserved because early NKT and IL-4 gene signatures also positively correlate with the levels of neutralizing antibodies in Zika-virus-infected macaques. In conclusion, our data support a model wherein a pre-TfH wave of IL-4 secreted by interfollicular NKT cells triggers the seeding of germinal center cells and serves as an innate link between viral infection and B cell immunity.
Graphical abstract Teaser NKT cells are required for the initial formation of germinal centers and production of effective neutralizing antibody responses against viruses.
Two immunoglobulin (Ig) diversification mechanisms collaborate to provide protective humoral immunity. Combinatorial assembly of IgH and IgL V region exons from gene segments generates preimmune Ig repertoires, expressed as B cell receptors (BCRs). Secondary diversification occurs when Ig V regions undergo somatic hypermutation (SHM) and affinity-based selection toward antigen in activated germinal center (GC) B cells. Secondary diversification is thought to only ripen the antigen-binding affinity of Igs that already exist (i.e., cognate Igs) because of chance generation during preimmune Ig diversification. However, whether stochastic activation of noncognate B cells can generate new affinity to antigen in GCs is unclear. Using a mouse model whose knock-in BCR does not functionally engage with immunizing antigen, we found that chronic immunization induced antigen-specific serological responses with diverse SHM-mediated antibody affinity maturation pathways and divergent epitope targeting. Thus, intrinsic GC B cell flexibility allows for somatic, noncognate B cell evolution, permitting de novo antigen recognition and subsequent antibody affinity maturation without initial preimmune BCR engagement.
Germinal centers (GCs) are micro-domains where B cells mature to develop high affinity antibodies. Inside a GC, B cells compete for antigen and T cell help, and the successful ones continue to evolve. New experimental results suggest that, under identical conditions, a wide spectrum of clonal diversity is observed in different GCs, and high affinity B cells are not always the ones selected. We use a birth, death and mutation model to study clonal competition in a GC over time. We find that, like all evolutionary processes, diversity loss is inherently stochastic. We study two selection mechanisms, birth-limited and death limited selection. While death limited selection maintains diversity and allows for slow clonal homogenization as affinity increases, birth limited selection results in more rapid takeover of successful clones. Finally, we qualitatively compare our model to experimental observations of clonal selection in mice.
Selection and maturation of B cells into plasma cells producing high-affinity antibodies occur in germinal centers (GC). GCs form transiently in secondary lymphoid organs upon antigen challenge, and the GC reaction is a highly regulated process. TGF-β is a potent negative regulator, but the influence of other family members including bone morphogenetic proteins (BMPs) is less known. Studies of human peripheral blood B lymphocytes showed that BMP-6 suppressed plasmablast differentiation, whereas BMP-7 induced apoptosis. Here, we show that human naïve and GC B cells had a strikingly different receptor expression pattern. GC B cells expressed high levels of BMP type I receptor but low levels of type II receptors, whereas naïve B cells had the opposite pattern. Furthermore, GC B cells had elevated levels of downstream signaling components SMAD1 and SMAD5, but reduced levels of the inhibitory SMAD7. Functional assays of GC B cells revealed that BMP-7 suppressed the viability-promoting effect of CD40L and IL-21, but had no effect on CD40L- and IL-21-induced differentiation into plasmablasts. BMP-7-induced apoptosis was counteracted by a selective TGF-β type I receptor (ALK4/5/7) inhibitor, but not by a selective BMP receptor type I inhibitor. Furthermore, overexpression of truncated ALK5 in a B-cell line counteracted BMP-7-induced apoptosis, whereas overexpression of truncated ALK4 had no effect. BMP-7 mRNA and protein was readily detected in tonsillar B cells, indicating a physiological relevance of the study. Altogether, we identified BMP-7 as a negative regulator of GC B-cell survival. The effect was counteracted by truncated ALK5, suggesting greater complexity in regulating BMP-7 signaling than previously believed.
|
The tumor necrosis factor receptor superfamily member HVEM is one of the most frequently mutated surface proteins in germinal center (GC)-derived B ce…...
Via Krishan Maggon
Research ArticleImmunology Free access | 10.1172/jci.insight.126732 Differential transcriptome and development of human peripheral plasma cell subsets Swetha Garimilla,1 Doan C. Nguyen,2 Jessica L. Halliley,3 Christopher Tipton,4,5 Alexander F. Rosenberg,6 Christopher F. Fucile,7 Celia L. Saney,2 Shuya Kyu,2 Denise Kaminski,8 Yu Qian,9 Richard H. Scheuermann,9 Greg Gibson,1 Iñaki Sanz,4,5 and F. Eun-Hyung Lee2,5 First published April 2, 2019 - More info Abstract Human antibody-secreting cells (ASCs) triggered by immunization are globally recognized as CD19loCD38hiCD27hi. Yet, different vaccines give rise to antibody responses of different longevity, suggesting ASC populations are heterogeneous. We define circulating-ASC heterogeneity in vaccine responses using multicolor flow cytometry, morphology, VH repertoire, and RNA transcriptome analysis. We also tested differential survival using a human cell-free system that mimics the bone marrow (BM) microniche. In peripheral blood, we identified 3 CD19+ and 2 CD19– ASC subsets. All subsets contributed to the vaccine-specific responses and were characterized by in vivo proliferation and activation. The VH repertoire demonstrated strong oligoclonality with extensive interconnectivity among the 5 subsets and switched memory B cells. Transcriptome analysis showed separation of CD19+ and CD19– subsets that included pathways such as cell cycle, hypoxia, TNF-α, and unfolded protein response. They also demonstrated similar long-term in vitro survival after 48 days. In summary, vaccine-induced ASCs with different surface markers (CD19 and CD138) are derived from shared proliferative precursors yet express distinctive transcriptomes. Equal survival indicates that all ASC compartments are endowed with long-lived potential. Accordingly, in vivo survival of peripheral long-lived plasma cells may be determined in part by their homing and residence in the BM microniche. Graphical Abstract Introduction High-affinity IgG and IgA antibodies provide serological memory that affords protection against previously encountered pathogens. The serologic protection is mediated by long-lived plasma cells (LLPCs), which have been identified in human bone marrow (BM) and the gastrointestinal tract (1–3). Initial response to vaccination is mounted by proliferative antibody-secreting cells (ASCs), which are highly enriched for antigen-specific cells that undergo a massive expansion for approximately 5–14 days after immunization (4–7). Yet, a fundamental gap in understanding remains regarding whether intrinsic programs of the ASC or extrinsic environmental factors determine survival to become an LLPC. The classic ASC population in blood is based on the relative expression of CD38 and CD27 on CD19+ cells. However, heterogeneity of the circulating ASC populations has been described extensively (8–12). Characterization of the CD19+ ASCs has recognized both CD138+ and CD138– populations in the blood after vaccination (4, 9, 12–14) and during steady state (12). Additional markers such as HLA-DR, Ki-67, CD95, and CD126 demonstrate recent activation of the ASCs in the blood after immunization (13). However, by focusing only on CD19+ ASCs (after excluding CD20+ cells), the complexity of blood ASC subsets in healthy vaccine responses would not have considered the CD19– ASC populations that resemble LLPCs (1). In autoimmune patients, the CD19– ASCs appear in the blood of diseased patients during flares (15), and recently, CD19–ASCs were also described after vaccination of healthy adults (14). Interestingly, contrary to proposed models of the release of old PCs from BM microniches, the CD19– ASC subsets in the blood were shown to have a fraction of new ASCs generated in response to vaccination (14). The identification of CD19–CD38hiCD138+ LLPCs (1) suggests that unique surface markers may play a role in maintaining survival. For example, CD138 was shown to play a direct role in PC survival in mouse models (16). By contrast, the role of CXCR4 in long-lived survival may be related to BM homing rather than intrinsic mechanisms (11). Additionally, loss of markers such as CD19, HLA-DR, and BCR may play a role in survival, although there is little evidence for this observation. Another interpretation for the loss of some markers may actually reflect distinct changes in the intracellular pathways such as G2M checkpoints, metabolism, apoptosis, and autophagy that have been described to sustain LLPCs (1, 17, 18). Nonetheless, it is unclear whether the unique surface markers on heterogeneous ASC populations signify intrinsic differences in cell survival programs. Germinal center responses play a crucial role in LLPC generation. It is thus possible that specific blood ASCs are imprinted during priming in the germinal center by the local milieu consisting of IL-21 from T follicular helper (Tfh) cells, follicular dendritic cells, and other T cell help (19–23). Thus, ASC heterogeneity may have evolved to distinguish particular ASC subsets with unique intrinsic mechanisms that are programmed to become long-lived. In addition to intrinsic mechanisms, extrinsic factors appear to play a critical role in LLPC survival. The BM survival niche plays an important role in the maintenance of LLPCs. The specialized niche that consists of hypoxia, secreted factors from the BM mesenchymal stromal cells (MSCs), and the cytokine APRIL, has recently been shown to maintain human ASCs for over 50 days in culture (24). Whether this environment actually changes the phenotype of the peripheral circulating blood ASCs into LLPCs or merely provides survival factors is still unclear. In this study, we used FLOw Clustering without K (FLOCK), an automated flow cytometry analysis program (4), to identify 5 distinct populations of ASCs that can be consistently isolated from human blood. Our data validate 3 CD19+ and 2 newly described CD19– ASC populations after vaccination. We also show that the majority of circulating CD138+ ASCs (both CD19+ and CD19–) are active participants in new vaccine responses and have undergone recent proliferation. Next-generation sequencing (NGS) analysis of the VH repertoire shows oligoclonality with a large degree of interconnectivity among the 5 subsets, and despite unique RNA signatures distinguishing populations 2 and 3 (CD19+) from population 5 (CD19–), those 3 populations have similar long-lived survival potential. Results Heterogeneity of human ASC subsets in blood. Human antibody responses after vaccination strongly correlate with a transient increase in circulating ASCs characterized by a CD19+CD27hiCD38+ phenotype (4, 25–28). However, the exact contribution of such cells to the long-lived protective antibody production is unclear, in part due to incomplete characterization of the ASC response and over-reliance on the expression of CD19. To address these questions, we performed PBMC fractionation of CD19+ and CD19– populations within CD3–CD14– cells at the peak of the ASC response after tetanus toxoid and influenza vaccination (6–7 days after immunization) (6). Interestingly, CD19– fractions were detected, albeit smaller in frequency than the CD19+ fractions (Figure 1A). Given the high frequency of vaccine-specific ASCs in both fractions, CD19+ and CD19– cells were analyzed using flow cytometry, yielding 5 putative ASC subsets distinguished by their relative expression of CD38 and CD138; these are population (pop) 1 (CD19+CD38+CD138–), pop 2 (CD19+CD38hiCD138–), pop 3 (CD19+CD38hiCD138+), pop 4 (CD19–CD38hiCD138–), and pop 5 (CD19–CD38hiCD138+) (Figure 1A and Table 1). Figure 1 ASC subsets in human blood 7 days after tetanus vaccination. (A) Top panels divide the CD19+ and CD19– fractions. Lower panels represent subsets of CD19–IgD– (left) and CD19+IgD– (right) fractions. (B) Morphology of blood ASC subsets (×100 magnification) by Wright-Giemsa stain. Left column: Sorted blood ASC subsets on day 7 after tetanus vaccination. ASC populations (pops) 1 to 5 and naive B cells are shown. Right column: Percentage of intracellular BLIMP-1 staining per subset is shown in blue histograms (naive controls in red). (C) Percentage of each ASC subset and naive B cells (N) expressing IgG, IgA, or IgM isotypes after peak vaccination. (D) Quantification of each blood ASC subset (pops 1 to 5) in cells/ml (top) and percentage of PBMCs (bottom). (E) Quantitative RNA expression of 5,000 sorted ASC subsets and naive and memory B cells for Pax5 (top), BLIMP-1 (middle), and Xbp-1 (lower), normalized to GAPDH in blood. Relative mRNA expression is expressed in arbitrary units. Table 1 Phenotype of blood ASC subsets 1 to 5 (number, %) We also employed an unbiased automated flow-gating program, FLOCK, with CD19+ and CD19– fractions, and validated these 5 subsets. FLOCK clusters cells in multidimensional hyperdense regions for each of the markers and separates cells into different subsets if they differ from other clusters in at least one marker/dimension. Previously, we identified pop 1 within the CD19+ fractions using FLOCK (4), although it is a relatively minor fraction of the CD19+ ASCs. This unsupervised approach revealed no additional ASC subsets for 6 subjects (Supplemental Figure 1 and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.126732DS1), demonstrating that our characterization using CD19, CD38, and CD138 is useful and consistent for elaborating the cellular basis of humoral immune responses. Morphology and expression of PC transcription factors. The nature of the ASC subsets identified by flow cytometry was validated by multiple approaches including morphology, expression of transcription factors involved in PC differentiation (BLIMP-1 and Xbp-1), and spontaneous antibody secretion. Morphology was ascertained by cytospin with Wright-Giemsa staining of each sorted ASC subset present in the blood after tetanus vaccination of 2 subjects (ages 50 and 59 years). Pops 1 to 5 had ASC characteristics including large size with big, eccentric nuclei and enlarged cytoplasm, distinct nucleoli, and a prominent Golgi zone (Figure 1B). Nearly all cells in pops 2 to 5 had intracellular BLIMP-1 protein detected by flow cytometry (Figure 1B). Interestingly, IgG isotypes were highest in ASC pops 2 to 5 except for pop 1 (Figure 1C). None were observed in naive B cells. Pop 1 had variable expression of BLIMP-1, suggesting it comprises an early or mixed population of ASCs. Pops 2 and 3 (CD19+ subsets) made up the largest frequencies of all the blood ASC subsets, and pops 4 and 5 (CD19– subsets) were the least abundant (Figure 1D). Additionally, pop 4 was quite heterogeneous; thus, additional backgating of the BLIMP-1–positive pop 4 cells showed no difference in forward or side scatter compared to other ASC populations (Supplemental Figure 2). All peripheral blood ASC populations (pops 1 to 5) were characterized by RNA expression of BLIMP-1 and Xbp-1 and absence of Pax5 (Figure 1E). In contrast, Pax5 expression was higher in naive versus memory B cells, neither of which expressed Xbp-1. BLIMP-1 was undetectable in memory B cells. The lack of Pax5 expression and increased BLIMP-1 expression in pop 1 suggests a process of differentiation from a B cell into an ASC. Xbp-1 is downstream of BLIMP-1 expression; thus, lower Xbp-1 expression observed in pop 1 compared with pops 2 to 5 also suggests that pop 1 may represent an earlier stage in ASC differentiation, or a mixed population of activated B cells that have already downregulated Pax5. A low level of BLIMP-1 expression was observed in naive B cells, which may have been due to a fraction of activated naive cells, or a small number of contaminating B1 cells, since only IgD and CD27 were used to discriminate the naive population, noting that a low level of BLIMP-1 expression has been reported in mouse B1 cells (29). Flow characteristics of blood ASC subsets. As B cells differentiate into ASCs, they undergo massive proliferation and lose features of B cells (such as CD20 and surface Ig), exit cell cycle, gain expression of BLIMP-1, and upregulate receptors for homing to the BM (30–32). Accordingly, we used multiparameter flow cytometry to evaluate these characteristics (Figure 2 and Table 1). As shown in Figure 2A, CD20 was downregulated on nearly every cell in pops 2 to 5, consistent with the loss of CD20 that begins as early as 7 days after vaccination in newly formed ASCs (4). However, pop 1 contained a mixture of cells positive for CD20, suggesting it is the earliest ASC phenotype with downregulation of Pax5 (Figure 1D). In contrast, surface Ig was highest on pop 1 and gradually downregulated in pops 2, 3, and 5 (~34%–45%; only ~8% in pop 4). These proportions of blood ASCs with surface Ig were higher than those found on LLPC (pop D) BM subsets (1). Interestingly, circulating ASC pops 2, 3, and 5 uniformly expressed high levels of CD27, a member of the tumor necrosis factor receptor (TNFR) family that is upregulated during B cell activation and is linked to PC differentiation (33), whereas the lowest frequencies of CD27 expression were found in pop 1 (~17%) (Figure 2A). Figure 2 Phenotype of blood ASC subsets on day 7 after vaccination. (A) CD20, surface Ig (kappa and lamda), and CD27 staining for blood ASC subsets and naive B cells (CD19+IgD+CD27–) illustrated in blue relative to controls in gray (also shown in right-hand panel). (B) HLA-DR and Ki-67 staining for blood ASC subsets. Far right: CD14+ peripheral blood monocytes served as controls for HLA-DR staining and naive B cells for Ki-67. (C and D) Frequency of CXCR4, CD28, IL-6R, FCGR2B, and BCMA in blood ASC subsets (pops 2 to 5) and naive B cells. Respective numbers of subjects are listed in Table 1. HLA-DR, a marker of cell activation previously shown to decrease during PC maturation (9), was highly expressed on nearly all blood ASC subsets (Figure 2B), which contrasts with its near absence on LLPC BM PC subsets (1, 2). We also found that Ki-67, a nuclear protein associated with recent cell division, was expressed by most cells in pops 2 to 5 and at a lower frequency of cells in pop 1 (Figure 2B). Thus, nearly all postvaccination ASCs in peripheral blood expressed Ki-67, consistent with the idea that these cells were recently generated, again in direct contrast to BM PCs (1). The relative expression of molecules involved in LLPC homing and survival were evaluated on blood ASCs (Figure 2C). For example, CXCR4, a chemokine receptor implicated in PC homing and retention in the BM (32, 34), was most commonly expressed in pop 1 and gradually decreased proportionately in pops 2 through 5 (Figure 2C). CD28, a costimulatory molecule associated with LLPC survival (35), which was known to have approximately 20% expression on BM LLPCs (1), was virtually absent on any of the blood ASC subsets. We previously reported that the IL-6R expression was very low on BM PC subsets despite the important role of IL-6 in PC survival (36). However, in the blood, ASC pops 3 to 5 expressed high levels of IL-6R (Figure 2C). Finally, the expression of the inhibitory FCGR2B, which promotes PC apoptosis (37), showed wide variation from subject to subject, but was typically low in blood ASC subsets compared with BM subsets (1) (Figure 2D). Lastly, B cell maturation antigen (BCMA), also known as TNFR superfamily member 17 (TNFRSF17), the receptor for APRIL, was uniformly expressed on pops 2–5 (Figure 2D), demonstrating the importance of this cytokine. Together, these results demonstrate that blood ASC subsets are Ki-67+ and HLA-DR+, suggestive of recent proliferation and activation. ASC subsets before and after vaccination in blood. Overall, compared with day 0 (~0.5%), we found that ASCs represented a larger fraction of circulating PBMCs 7 days after vaccination but with variable input from the different populations, as CD19+ (pops 1, 2, and 3) contributed in excess of 95%. This frequency is in keeping with the postvaccination levels of circulating ASCs previously established by enzyme-linked immunospot (ELISpot) analysis, which typically represent a 5- to 20-fold increase over steady-state levels (28). Therefore, we sought to determine the relative contributions of the different ASC subsets to this substantial increase after tetanus vaccination. We performed multicolor flow cytometry in 8 healthy adults prior to (i.e., steady state) and at peak vaccine responses. We removed pop 1 due to its low abundance and heterogeneity. The average total number of ASCs in each population increased after vaccination as follows: pop 2 increased by 7.6-fold, pop 3 increased 33-fold, and pop 5 increased by 13-fold. Thus, the relative abundance at steady-state and peak vaccine responses were different. During steady state, pop 2 (~60%–80%) dominated the ASC ratios with almost no pop 5; however, vaccination induced relative increases of expansions of pops 3, 4, and 5 in all 8 subjects, with corresponding decreases in pop 2 proportionally (Figure 3A). This appearance of CD19+CD138+ fractions (pop 3) for healthy adults after immunization is consistent with previous observations after tetanus vaccination (13). Figure 3 Ratios of ASC subsets in blood and BM. (A) Pie charts representing proportions of pops 2, 3, 4, and 5 in the blood from 8 different adult subjects at steady state (top) and at peak (days 6–7) ASC response in blood after tetanus vaccination. The proportion of each ASC subset is represented by the corresponding sector size of the pie chart. Kinetics of the ratios of the ASC in the blood after tetanus vaccination is shown for subject 3 (inset). (B) From 8 additional subjects, ratios of pops A, B, C, and D in the BM and blood pops 2, 3, 4, and 5 were matched at the time of the BM aspirate. ASC subsets in blood and BM (pops 1 and Z) are not included. Pops 2 and A, pops 3 and B, pops 4 and C, and pops 5 and D (LLPC subset) are shown in tan, red, blue, and green, respectively. Frequencies of ASC subsets in blood and BM. Direct comparison of the quality and quantity of the ASC subsets in 8 healthy asymptomatic adults (mean age 48.5, range 43 to 56 years) was also performed for matched blood and BM samples from the same subject. Pop A in the BM, which contains surface markers similar to those in ASC pop 2 in the blood, make up a markedly smaller percentage of BM ASC subsets compared with the blood (Figure 3B). The relative frequencies of the different ASCs were remarkably conserved in all 8 BM samples analyzed, with CD138+ (pops B and D) representing the most abundant subsets. Pop 5 could be detected in the blood in some subjects at steady state, but most of these cells were positive by Ki-67 staining (Figure 2B), thereby distinguishing them from BM pop D (Ki-67–) with the same surface markers (1). We found that at steady state, pop 5 had similar Ki-67+ staining that resembled the blood pop 5 during acute immune responses, implying that they are newly generated ASCs and not BM emigrants released in the blood from the BM niches. NGS of the VH antibody repertoire of ASC subsets. NGS was used to assess the complexity and connectivity of the antibody repertoire expressed by the different ASC subsets. Pops 1 to 5 were sorted from a 45-year-old adult after tetanus vaccination and 3,174; 5,000; 5,669; 2,550; and 1,994 cells, respectively, were collected. NGS was performed using Illumina MiSeq Amplicon sequencing with primers positioned in the framework region FR1 of the VH1–VH6 families, and in constant regions corresponding to the IgM, IgG, and IgA isotypes (Figure 4). Sequences were considered part of the same clonal lineage if they shared the same V and J gene segment rearrangement and a CDR3-H of identical length with at least 85% sequence similarity based on our previous studies (38). Total numbers of lineages for pops 1–5 were 984, 943, 825, 83, and 311, respectively. The overall degree of clonality was quantified using D50 or D20 scores calculated as the number of clonotypes accounting for the top 50% or 20%, respectively, of all size-ranked clones within a given population. For ASC pops 1 to 5 and memory B cells, the IgG D20 scores were 14, 2, 2, 1, 2, and 2, and D50 scores were 65, 4, 6, 3, 5, and 9, respectively, demonstrating that each subset at the peak of the vaccine response is dominated by a small number of substantially expanded ASC clones (Figure 4A). Switched memory (SWM) B cells were also oligoclonal, whereas the naive B cells were polyclonal with D20 and D50 scores of 910 and 3,674, respectively. The diversity of the VH-gene repertoire is demonstrated by the Hill diversity score and was highest in pop 1 compared with the other ASC subsets (Figure 4B). Finally, the composition of the isotypes of the VH sequences in the ASC subsets (Figure 4C) were switched between IgG and IgA sequences, with predominantly IgA compared with IgG in pop 1, whereas pops 2 to 5 and SWM B cells were higher in IgG. Very little IgM isotypes were found in the ASC subsets or SWM B cells, whereas naive B cells contained only IgM sequences, as expected. Figure 4 Next-generation sequencing (NGS) repertoire sequencing of blood ASC subsets. NGS was used to analyze the clonal repertoire of the ASC populations, naive B cells, and isotype-switched memory B cells. (A) Diversity of the repertoire is shown by plotting lineage (clone) size versus the cumulative percentage of sequences determined from size-ranked clones. Largest clones are found at the top of the plot and account for a greater area within the subdivided plots. More diverse repertoires, such as the naive population here, only contain small clones in a more even representation. (B) Hill diversity profiles for each population (with different levels of sampling) demonstrate the overall diversity of the repertoire in each of the ASC populations. (C) Relative quantities of IgM, IgG, and IgA sequences in each subset. Naive is predominantly IgM. Blood ASC subsets 1 to 5 and switched memory B cells show mostly IgG and IgA. (D) Circos plot shows interconnectedness of the ASC populations by plotting the sequences from each population in clonal size–ranked order, with the largest clones being in the most clockwise portion of each population segment. The outer-most track shows the isotype makeup of each clone by color. The next track in shows mutation frequency of each sequence, with more mutations represented as more distal from the center of the plot. The next track in shows the number of sequences, followed by the clonality displayed by a circular stacked bar plot. Here, only the largest 50% of the clones are colored to avoid blurring of small clones. The internal connections show clones found in multiple populations. (E) Stacked bar plots again demonstrate the diversity of the repertoire by showing size-ranked clones as segments taking up a percentage of the total repertoire. The largest 10 clones of all populations are colored and like-colors demonstrate the same clone in multiple populations. (F) The Morisita overlap index demonstrates the similarity of repertoires in various populations as a value from 0 (no similarity) to 1 (identical repertoires). The color strength is indicative of interconnectivity. Next, the actively expanded blood IgG, IgA, and IgM ASCs in response to immunization shared similar repertoires, as shown in the outer circos plot tracks (Figure 4D). Connectivity of the repertoires among subsets is also shown by the circos plot (39) (Figure 4D). Blood ASCs were largely oligoclonal with a repertoire dominated by a few clonotypes, many of which in ASC pops 2 to 5 were shared (Figures 4, D and E). The SWM B cells were also highly oligoclonal with many shared clones, in contrast to the naive B cell subset. Many of these same clones were also shared by ASCs in pop 1, but consisted of much smaller clonal populations (Figure 4E). Pops 1 to 5, as well as SWM B cells, were all highly interconnected as quantified by the high Morisita overlap indices (Figure 4F), but this was not the case comparing ASCs with naive B cells. In conclusion, the VH repertoire analysis demonstrates oligoclonality, rich interconnections, and predominately IgG isotypes among the circulating ASC subsets and SWM B cells on day 7. Mutation analysis and VH lineage analyses. On average, all blood ASC pops 1 to 5, which were mostly class switched to IgG or IgA, were highly mutated with similar average mutation frequency, as defined by the number of mutations in each sequence divided by length of that sequence (Supplemental Figure 3A). Intraclonal mutation analysis was also conducted to determine progression of ASC populations within individual clones. In this analysis, average mutation frequencies for each individual population were compared to average mutation frequencies of all populations in that same clone. By examining the mutation frequencies this way, we can get a better picture of which populations tend to have higher or lower mutation frequencies compared with other populations within the same clone. This analysis showed some deviations of the individual ASC populations’ mutation frequencies, namely that pop 4 tended to have the highest mutation frequencies in clones, while SWM and pop 2 tended to have the lowest (Supplemental Figure 3B). To follow the possibility of sequential acquisition of mutations for the different subsets, we analyzed the VH repertoire of 5 of the largest individual clones that shared lineages in pops 1 to 5 using IgTree (40) and PHYLIP phylogenetic analysis. Within the 5 clones, the number of nodes shared among the various populations of ASCs was striking. No apparent progression or sequential differentiation was found, but instead the ASC populations were highly intermixed throughout the phylogenetic tree. An example of one clone shows a deep IgTree structure with high interconnectedness between populations, illustrating the single origin of the multiple blood ASC subsets (Supplemental Figure 3C). Thus, we concluded that sequential increases in progressive accumulation of mutation did not occur from pops 1, 2, 3, 4, and 5, but each clone segregated into independent branches, demonstrating that both CD19+ and CD19– subsets arise from a common B cell progenitor. Tetanus IgG secretion in blood ASC subsets. The secretory function of different ASC subsets was validated by measurement of constitutive antibody secretion as well as participation in antigen-specific responses after tetanus vaccination. Spontaneous total IgG and tetanus-specific IgG without in vitro stimulation were assessed by ELISpot assays in 6 adults (mean age 44 ± 11 years, range 27–59 years) from blood after tetanus boosting. Cells constitutively secreting IgG were detected in all 5 ASC subsets but not every subject had adequate numbers of pops 4 and 5 for FACS analysis. Nonetheless, tetanus-specific IgG ASCs were highly enriched in both CD19+ (pops 1, 2, and 3) and CD19– (pops 4 and 5) ASC fractions when present (mean 19%, 29%, 33%, 33%, and 25% for pops 1 to 5, respectively) (Figure 5, A and B). Thus, all circulating ASCs (including the CD19– and CD138+ subsets) can contribute similarly to the short-term response to tetanus immunization. Figure 5 Vaccine-specific IgG ASC frequencies 7 days after tetanus vaccination. (A) ELISpot of total IgG (top) and tetanus-specific IgG (lower) ASC frequencies from sorted ASC subsets (pops 1 to 5) in the blood. Naive B cells and total PBMCs are also shown. The total number of sorted cells per well is indicated adjacent to each well. (B) Percentage of tetanus-specific IgG/total IgG ASC frequencies from sorted ASC subsets (pops 1–5) in blood ASC populations from 8 adults. Note: some patients had limited frequencies of pops 4 and 5, which could not be sorted. Comparative transcriptome analysis of 3 ASC subsets. The transcriptomes of pops 2, 3, and 5 were contrasted by analysis of variance of RNA sequencing (RNA-seq) profiles of 6 donors (labeled Sub 06 to 11). Two-way hierarchical clustering of 674 genes differentially expressed among the 3 ASC pops shows that pop 5 is significantly different from pops 2 and 3 (Figure 6A). This basic distinction between the CD19– and CD19+ ASCs is supported by principal component analysis (PCA), since PC1, which captures 56% of the gene expression variance, separates pops 2 and 3 from pop 5, while PC2 (7%) distinguishes pop 2 and pop 3 (Supplemental Figure 4). Furthermore, the 464 genes upregulated in both pops 2 and 3, and 210 genes downregulated relative to pop 5, lead to coclustering of the pairs of samples within individuals, implying that the 2 subtypes are transcriptionally closely related within subjects. Figure 6 Transcriptomic analysis of ASC subsets. (A) Heatmap of 672 transcripts differentially expressed among pop 2, pop 3, and pop 5 RNA-seq profiles from 6 subjects. Red color indicates relatively high expression, blue low expression. Two major blocks of genes upregulated or downregulated in pop 5 relative to pops 2 and 3 are indicated. Sample identities to the right show that pairs of samples from the same individual tend to cluster together. (B) Heatmap of PC1 of 29 gene sets found to be enriched in the differentially expressed genes also shows differentiation of pop 5 from pops 2 and 3, with only minor differences between the latter two. (C) Bubble plots show significantly biased gene sets in each pairwise comparison, with the size of the bubble proportional to the negative logarithm of the P value for the normalized enrichment score indicated along the x axis (see Supplemental Table 2 for full list of pathway names). Gene set enrichment analysis (GSEA) reinforces this similarity between pops 2 and 3 in comparison with pop 5 and highlights 29 gene sets that are differentially regulated. These gene sets summarize biological functions that are likely to differ between the pops, and are visualized in 3 different ways since raw summary statistics can misrepresent the relationship between upregulation of transcription and pathway activity. In Figure 6B (and Supplemental Figure 5), the first principal component, which in all cases explains over 45% of the variance in the gene set and has been polarized to ensure that positive values represent a preponderance of upregulated transcripts in the gene set, suggests upregulated pathways of cellular metabolism (adipogenesis, glycolysis, oxidative phosphorylation, fatty acid metabolism, and mTORC1 signaling), stress-induced pathways (DNA repair, UV response, and unfolded protein response), and cell cycle pathways (E2F targets, Myc targets, and G2M checkpoints) in pops 2 and 3. By contrast, several signaling pathways (JAK-STAT3, PI3K-AKT, IFN response, TGF-β signaling, and TNF-α signaling) as well as the hypoxic response appear to be elevated in pop 5. Figure 6C reinforces most of these conclusions by presenting the results of normalized enrichments scores for each pathway, but further reveals a gradient whereby pop 2 is more extreme than pop 3 for several pathways, namely downregulation of TNF-α signaling and upregulation of Myc and E2F target. G2M checkpoint regulators decreased expression with acquisition of CD138 and loss of CD19. This difference in dysregulated pathways suggests different cellular functions for pops 2 and 3 in comparison with pop 5. To visualize differentially expressed genes contributing to pathways, we used spider plots (Figure 7) to contrast the directions of all genes in a gene set whose transcript abundance significantly differed between 2 or more of the ASC subsets. We included some gene sets from which the hallmark pathways are derived and which were of a priori interest. For example, for apoptosis (contained within allograft rejection), hypoxia, TNF-α signaling, and the cell cycle (E2F targets, G2M checkpoints), pop 5 shows very clear upregulation of specific genes resulting in a green polygon that is more expanded along the arcs of the web, whereas the blue-colored pop 3 and pop 2 differential expression is more similar to one another, producing overlapping polygons. This analysis also indicates reduction in expression of all or most of the indicated genes encoding extracellular matrix (ECM) or unfolded protein response (UPR) functions in pop 5. It is not, however, simple to extrapolate transcript abundance to biological functions. For example, the hypoxic response engages 6 genes involved in autophagy; 3 inhibitors of the process (LDHA, PRKCB, and CYBB) are elevated in pop 2, whereas another, MAPK3, is elevated in pop 5, while PIK3CD and MTOR also regulate ER stress and autophagy, yet are expressed in opposite directions. Prediction of the consequences of differential expression awaits systems modeling that is sensitive to the precise nature of which genes are up- or downregulated jointly. Figure 7 Significantly differentially expressed genes in selected pathways. Spider plots of significantly differentially expressed genes in 6 selected pathways showing differences among the 3 ASC populations. Rays of each plot represent transcript abundance for the indicated gene, with low values in the center and high at the periphery. Polygons link observed transcript levels in each cell type, showing how pop 5 differs from pops 2 and 3. ASC survival potential in a human in vitro BM culture system. To discriminate the differential survival potential upon arrival in the BM microniche, we developed an in vitro cell-free culture system that mimics the human BM microenvironment, as previously described (24). For proof of concept using one abundant blood ASC pop 3, we performed long-term cultures and compared survival for 50 days in the BM MSC secretome alone or secretome with the addition of exogenous APRIL in normoxic or hypoxic conditions from a patient (age 23 years) 7 days after hepatitis A vaccination (Figure 8A). Pop 3 survival was barely maintained by day 50 in the secretome alone in normoxia, but a fraction of cells had better survival in secretome and hypoxia. Interestingly, survival was best overall with the secretome with the addition of exogenous APRIL in hypoxic conditions (Figure 8B). With optimal conditions for ASC survival similar to conditions previously shown with total blood ASCs (24), we sorted ASC pops 2, 3, and 5 from 2 subjects (38 and 25 years old) after tetanus toxoid or influenza vaccination, and cultured them in secretome with the addition of exogenous APRIL in hypoxia for 21 or 48 days, respectively (Figures 8, C and D). The low abundance and heterogeneity of pop 4 made them difficult to culture and maintain. The ASC subsets (pops 2, 3, and 5) were easily sustained in culture and pop 2 may have had a slight survival advantage earlier in culture (days 14–21); however, by day 48, pops 2, 3, and 5 had similar survival rates. Thus, both CD19+ (pops 2 and 3) and CD19– (pop 5) ASC subsets could be maintained in culture for nearly 2 months and all subsets have similar fractions of cells with LLPC potential. Figure 8 In vitro human BM microniche systems to measure long-lived survival of the subsets. (A) IgG ELISpots of pop 3 ASCs from a healthy adult after hepatitis A vaccine on days 0, 7, 35, and 50 in the BM MSC secretome (green) or BM MSC secretome with the addition of exogenous APRIL (red) in normoxia and hypoxia (blue open symbol or blue closed symbol). (B) Percentage of IgG ELISpots (relative to the maximal frequency) for pop 3 in MSC secretome in normoxia (green square), MSC secretome in hypoxia (open blue square), MSC secretome with the addition of exogenous APRIL in normoxia (red square), and MSC secretome with the addition of exogenous APRIL in hypoxia (blue square). (C) Percentage of IgG ELISpots from pops 2, 3, and 5 (relative to the maximal frequency for each population) from a healthy adult after tetanus vaccination on days 1, 7, and 21 in MSC secretome with the addition of exogenous APRIL in hypoxia. (D) Percentage of IgG ELISpots from pops 2, 3, and 5 (relative to the maximal frequency) from a healthy adult after influenza vaccination on days 1, 14, 28, and 48 in MSC secretome with the addition of exogenous APRIL in hypoxia. To understand if the ASC maturation required CD138 upregulation, we cultured FACS-isolated CD19+CD27hiCD38hi ASCs from a healthy adult in our BM mimic (MSC secretome, exogenous APRIL, and hypoxic conditions). We performed confocal staining of the ASCs before and 14 days after culture. We show an increased frequency of ASCs with CD138+ staining on day 14 (Supplemental Figure 6). Whether CD138– ASCs upregulated CD138 or CD138+ ASCs preferentially survived in the cultures will need additional studies. Nonetheless, increased frequencies of CD138+ ASCs are shown after culture in the BM microniche and are likely related to enhanced survival. Discussion LLPCs are the cornerstone of vaccinology and lifetime protective mediators of infection. It is widely accepted that during acute-recall immune responses, newly generated, proliferative ASCs with a short lifespan produce a transient burst of antigen-specific antibodies. After the initial response decays, pathogen-specific antibody production is sustained at lower levels by mature, nonproliferating, terminally differentiated PCs capable of surviving for many years in the BM (i.e., LLPCs) in the absence of additional antigenic stimulation. Whether all peripheral blood ASCs eventually evolve into LLPCs upon taking residence in BM survival niches, or if short-lived ASC and LLPC precursors are imprinted with a long lifespan based on the relative strength of antigenic signaling received by the corresponding precursor B cells, represent 2 distinct models (41). Discriminating between these 2 models is dependent on the ability to recognize different subsets of ASCs in the blood and their relationship with BM LLPCs. In this study, we demonstrate the heterogeneity of blood ASC subsets after vaccination by identifying both CD19+ and CD19– subsets with cell surface markers similar to BM PC subsets, as we have previously shown (1). Our results demonstrate significant heterogeneity of human ASC subsets in the blood after vaccination. These heterogeneous subsets include traditional plasmablasts (CD19+CD38hiCD27hi) and 4 newly defined ASC populations distinguished by their relative expression of the surface markers CD19, CD38, and CD138. Discovery and confirmation using FLOCK analysis (4) was essential to comprehensively identify all antibody-producing cells within these subsets. We also show that all circulating ASC subsets are active participants in recent vaccine/immune responses. Interestingly, peripheral CD19–CD138+ ASCs are universally proliferative (>90% Ki-67+) and contain antigen-specific responses with frequencies similar to conventional CD19+CD138– plasmablasts. Also, these antigen-specific ASCs with mature phenotypes are found in the periphery within a few days of immunization, arising at the same time as CD19+ ASCs. Most importantly, CD19– ASCs cannot be the result of displaced CD19– BM LLPCs, since BM LLPCs are almost universally Ki-67 negative (1). This conclusion is further supported by the high frequency of recent vaccine-specific cells and oligoclonality in the blood ASC subsets disclosed by VH repertoire studies within CD19– subsets. In contrast, VH repertoires of the BM PC subsets in healthy adults are highly polyclonal, representing an historical archive of a lifetime of exposures (1). Thus, the CD19–CD138+ ASC subsets in the blood are not displaced LLPCs from the BM microniche, but rather newer clones arising from the ongoing vaccine response. Composition of the antibody repertoire expressed by ASC and B cell subsets provides important clues regarding the origin, diversification, and selection of the compartments in question. Our results portray global repertoires expressed by multiple blood ASC subsets at the peak of vaccination, whereas previous studies had been limited to the analysis of a small number of randomly sampled single cells or global CD19+CD27hiCD38hi ASCs (instead of heterogeneous ASC subsets) (42, 43). The strong connectivity of the ASC subsets with the memory B cell fractions on day 7 may reflect the activated B cell phenotypes previously described as CD19+CD71+IgD–CD38lo/int (43). Consistent with single-cell studies, our global analyses demonstrate that responding ASCs were largely oligoclonal with highly variable rates of somatic hypermutation in the expanded clones, including some with rather low mutational load, a finding suggesting that some ASC clones might derive from recent differentiation of naive B cells. Nevertheless, the majority of expanded ASC clones were shared by all blood ASC populations and SWM B cells irrespective of the degree of mutation, which suggests they all derive from a common ancestral precursor B cell. Consistently, we also observe that, rather than forming a longitudinal gradient of progressive accumulation of mutation from pops 1 to 5 with shared VDJ clonotypes, individual clones within each ASC subset segregated into independent branches. These results indicate that all the circulating ASC subsets, including those with presumed mature phenotypes as indicated by the absence of CD19 and/or expression of CD138, derive from a common B cell progenitor. Then, they appear to develop through parallel differentiation of proliferating cells that acquire the characteristic cell surface phenotypes and divergent patterns of somatic hypermutation. Thus, ASC surface maturation (CD138 or loss of CD19) is likely independent of somatic hypermutation. Intriguingly, pop 1 had many IgA isotypes that predominated this subset with some connectivity to pops 2–5 and SWM B cells, albeit the least of all the subsets (Figure 4, A–F). Pop 1 ASCs were also the most diverse compared with the other ASC and SWM B cell subsets, suggesting that some ASCs in this population may originate differently. It is known that at steady state circulating ASCs are mainly IgA ASCs (44) (our unpublished data); however, the specificities remain unknown. Thus, pop 1 may contain both early vaccine-specific IgG ASCs and IgA ASCs from other sites. Whether they are homeostatic IgA ASCs specific to our microbial flora would be important to ascertain, but this will require further studies. Interestingly, pops 2, 3, and 5 had similar survival rates in our cell-free BM microniche system, implying equal survival potential upon arrival in a supportive niche in the body. Other models using in vitro differentiated ASCs had a massive attrition of ASCs in culture since they did not optimize conditions to mimic the human BM microniche that is the naturally occurring site of LLPC survival. Furthermore, they started with B cells that were differentiated to ASCs in vitro (14), compared with our studies that immediately cultured in vivo–differentiated ASCs, which is more physiologically relevant. Lastly, those models used MSC cell lines that are not likely to be as efficient as our validated human in vitro BM microniche system with primary BM MSC secretomes, exogenous cytokines, and hypoxic conditions to maintain human PCs (14, 24). Mouse models of LLPCs have been described in the BM demonstrating how homing and retention in the BM appears to be a major determinant for the persistence of human LLPCs (25). CXCR4 is important for PC migration and survival in the BM since BM stroma is rich in CXCL12 (45, 46). The CD19+ blood ASCs expressing higher CXCR4 levels may have potential for homing to supportive BM sites. At the same time, BM-resident LLPCs express very high levels of CXCR4, indicating that CXCL12 may also be involved in BM retention (1). Thus, with proper migration and retention, survival may be guaranteed given transcriptional programs intrinsic to the blood PCs. Since the BM-derived MSCs are an abundant source of CXCL12, whether CXCL12 also has a role in survival or just in retention will need further exploration. Our models could not discern a survival advantage of ASCs that may have intrinsic properties for BM homing because our system provides the same BM survival microniche equally to each sorted ASC subset (pops 2, 3, and 5). Therefore, the similar survival advantage of all 3 ASC subsets (Figure 8) may in fact reflect equal survival potential but not necessarily equal homing to or retention in the BM microniche. Recently, CD138, a heparin-sulfate glycoprotein (HSGP), has been shown to potentiate survival of ASCs (16). It was found that the ASCs lacking CD138 were more prone to apoptosis and reduced levels of IL-6 signaling. Evidence of increased ASC expression of surface HSGP is known to be important for the binding of survival factors, such as APRIL, hepatic growth factor (HGF), and epidermal growth factor (EGF), on malignant PCs (47–49); however, it is unclear if it is also important in normal LLPC development. More interestingly, IL-6 and APRIL protect ASCs from apoptosis in experiments using heparin sulfate chains to increase IL-6 and APRIL presentation on the ASC receptors (16). Although both CD138+ (pops 3 and 5) and CD138– (pop 2) ASC subsets had similar survival in our cultures, it was not clear if pop 2 also increased CD138 expression progressively within the in vitro BM microniche with higher concentrations of IL-6 and APRIL. These findings imply that the BM environment may play a role in altering the phenotype of an ASC through additional maturation. CD38+ ASC maturation appears to involve acquisition of CD138 in pop 3 (CD19+CD138+) and pop 5 (CD19–CD138+) cells, suggesting sequential maturation. B cells undergo massive proliferation and then differentiation to become ASCs (50). Our gene expression data indicated engagement of cell cycle, specifically G2M transition, genes among the various subsets, but it remains unclear whether pop 2 is the more proliferative subtype, as would be consistent with a maturation process that starts with pop 2 differentiating into pop 3. Furthermore, pops 2 and 3 showed elevated expression of caspase 3 and so are likely to undergo apoptosis, unless additional ASC maturation is orchestrated. Pop 5 ASCs are quite different from pops 2 and 3, with upregulation of hypoxia, TNF-α, and downregulation of UPR pathways. Because they have downregulated both oxidative phosphorylation and glycolysis, it will be interesting to understand the consequences for their survival, immune-related activity, and potential for ongoing maturation. Recent work by Neu et al. with single-cell transcriptomes of vaccine- versus non–vaccine-specific ASCs showed that the differences between these 2 subsets were in the glycosylation enzymes (51). These findings are interesting but address very different questions from our work. Our studies depict progression of ASC differentiation to a long-lived phenotype with the comparison of CD19+ and CD19– ASCs with upregulation of pathways such as hypoxia, TNF-α, and UPR involved in LLPC generation, whereas Neu et al. compared vaccine- versus non–vaccine-specific ASCs. Interestingly, the differences between vaccine- and non–vaccine-specific ASCs were relatively minor. One reason could be that similar transcriptomes of vaccine- versus non–vaccine-specific ASCs arise with short-lived (influenza) vaccines. Greater differences may have been observed between vaccine- and non–vaccine-specific ASCs in long-lived immunization such as tetanus. Nonetheless, additional studies are needed to further elucidate mechanisms of early ASCs to understand long-lived vaccine durability. Whether the local microenvironment is sufficient for LLPC generation is not known. In this study, we show that both CD19+ and CD19– ASCs have similar potential for longevity, suggesting that both extrinsic factors and intrinsic ASC programming may be essential. However, we also tested ASC subsets from vaccines with relatively long-lived protection such as tetanus (10 years), hepatitis A (25 years), and intermediate longevity for influenza vaccines (52, 53). We have not yet evaluated short-lived vaccines to test the imprinting models with differential strength of antigen signal, or costimulation with Tfh cells, to address this question. Hence, we show that the unique BM locale is necessary, but whether it is sufficient is still unclear. The improved survival in hypoxia within the BM microniche system was quite surprising. Clearly, the circulating ASCs in normoxic peripheral blood must adapt to hostile conditions of 2.5% oxygen. Whether these programs are intrinsic to the blood ASCs as they exit the germinal centers or whether they further differentiate to acclimatize to these microniches is not known. Moreover, mechanisms of how hypoxia provides a survival advantage will also need systematic evaluation of cultured ASCs. In conclusion, we find that significant heterogeneity of ASC subsets resides in the blood during active immune responses in healthy adults. Our results demonstrate that the timing of ASC subsets with markers of maturity (loss of CD19 and acquisition of CD138) are concurrent to CD19+ plasmablasts (CD19+CD27hiCD38hi) in the blood. Furthermore, both CD19+ and CD19– ASC subsets are newly generated, and all are participants of the new vaccine response. On aggregate, our results favor an ongoing evolution of ASCs once released into the peripheral blood and upon arrival to the BM niches. Thus, it is likely that the ASCs have intrinsic mechanisms of maturation (acquisition of CD138 and loss of B cells surface markers, such as CD19), which are further enhanced with extrinsic signals from the BM microniche such as IL-6 and APRIL. The surface surrogate markers may in fact be associated with important inflammatory mediators, such as IL-6, TNF-α, apoptotic pathways, along with metabolic pathway regulation to finally mature into LLPCs. Understanding these mechanisms will be important to study diseases of allergy, transplantation, and autoimmunity and to help develop better long-lived vaccines. Methods Subjects and study approval. Vaccinated and healthy asymptomatic adults (102 healthy adult subjects, 22 to 65 years old, mean 42 ± 11 years, 72 female, 30 male) were enrolled in this study at the University of Rochester Medical Center and Emory University during 2008–2017. Subjects received the tetanus toxoid Td or combination Tdap, influenza, or hepatits A vaccines as a part of routine medical care. PBMCs were isolated before vaccination, and on days 6–7 for all vaccination subjects. All studies were approved by the Institutional Review Boards at the University of Rochester Medical Center and at Emory University. Written informed consent from participants was obtained. BM aspirates. BM aspirates used for isolating ASC subsets were obtained from 8 healthy adults (43 to 56 years old, mean 49 ± 5 years, all female). Mononuclear cells were isolated by density gradient centrifugation. Peripheral blood was also collected from 8 subjects at the time of BM aspiration and PBMCs were isolated. Isolation and expansion of healthy BM-derived MSCs (from 2 unrelated healthy BM donors) and subsequent making of the MSC secretome were performed as previously described (24). Multicolor flow cytometry. PBMCs from peripheral blood or BM were isolated using a Ficoll density gradient and stained with the following anti-human antibody staining reagents: Ki-67–FITC (catalog, MHKI6701), CD3–PE-Cy5.5 (catalog, MHCD0318), CD14–PE-Cy5.5 (catalog, MHCD1418) (Invitrogen); CD20-Cy5 (catalog, 15-0209), CXCR4–PE-Cy5 (catalog, 15-9999), CD27–APC-eFluor 780 (catalog, 47-0279) (eBioscience); CD28-PE (BioLegend, catalog 302907); CD19–PE-Cy7 (catalog, 557835), IgD-PE (catalog, 555779), IL6R-PE (catalog, 561696), kappa- or lambda-PE (kappa, 555792; lambda, 555797), CD38–Pacific Blue (catalog, 561378), HLA-DR–Alexa Fluor 700 (catalog, 560743) (BD Pharmingen); CD138-APC (Miltenyi Biotec, catalog 130-091-250); and FCGR2B–Alexa Fluor 647 (custom conjugated by I. Sanz). The cells were analyzed on an LSRII flow cytometer (BD Biosciences). FLOCK analysis. FLOCK is a web-based program publically available for open use by the immunology research community through the Immunology Database and Analysis Portal—ImmPort (http://www.immport.org) (4). FLOCK is a novel multidimensional automated flow gating program that uses a density-based clustering approach to algorithmically identify cell populations from multiple samples in an unbiased fashion, thereby eliminating operator-dependent variability (4). FLOCK analysis was used for both total CD19+CD3–CD14– and CD19–CD3–CD14– PBMC populations from 6 blood samples to identify ASC subsets in an unsupervised fashion. These were subsequently isolated by sorting with cell-type specific antibodies. ASC subsets sorted by flow cytometry. CD3 and CD14 cells were removed by positive selection (CD3 and CD14 positive selection, Miltenyi Biotec; CD3, 130-050-101; CD14, 130-050-201) from the mononuclear cells isolated from blood or BM. CD3–CD14– cell fractions were stained with the following anti-human antibody staining reagents: IgD-PE (catalog, 555779), CD19–PE-Cy7 (catalog, 557835), CD38–Pacific Blue (catalog, 561378) (BD Pharmingen); CD3–PE-Cy5.5 (catalog MHCD0318), CD14–PE-Cy5.5 (catalog MHCD1418) (Invitrogen); CD138-APC (Miltenyi Biotec, catalog 130-091-250); and CD27–APC-eFluor 780 (eBioscience, catalog 47-0279). Naive and memory B cell fractions as well as multiple ASC subsets were collected (see Figure 1). Approximately 5,000 to 100,000 cells were collected for each population. Cytospins of sorted ASC subsets. Cytospins were performed from sorted ASCs in the blood at 1,300 rpm for 5 minutes on the Cytospin 4 (Thermo Fisher Scientific). Approximately 5,000 sorted cells per subset were dried overnight on albumin-coated slides and stained with Wright stain. Morphology was reviewed by a board certified pathologist and hematologist. In vitro culture systems for ASCs. In vitro cultures of human blood ASCs were performed as previously described (24). Briefly, ASCs were cultured in cell-free MSC secretome media in 96-well flat-bottom cell culture plates (Corning/Sigma) at 37°C in a humid, 5% CO2, 95% air (20% O2) incubator or in hypoxic culture conditions (2.5% O2) at 37°C in a modular incubator chamber (Billups-Rothenberg) that was infused with a preanalyzed gas mixture (AirGas) or in a cell culture incubator programmed for the desired O2 tension. The blood ASC survival and function were assessed by ELISpot assays, and their output values were expressed as the percentage of maximal IgG-secreting ASCs, which typically occurred on days 1–3. Total Ig and antigen-specific ELISpot assays. To assess survival and Ig secretion function of cultured ASCs, ELISpot assay was performed, as previously described (5, 6, 24, 28). Briefly, PBMCs, sorted ASCs, or B cell subsets were added to 96-well ELISpot plates coated with anti–human IgG (5 μg/ml, Jackson Immunoresearch), anti–human IgA (5 μg/ml, Jackson Immunoresearch), or tetanus toxoid (2 μg/ml, EMD Biosciences), and were incubated overnight. Wells were washed and bound antibodies were detected with alkaline phosphatase–conjugated anti–human IgG antibody (1 μg/ml, Jackson Immunoreseach), alkaline phosphatase–conjugated anti–human IgA antibody (1 μg/ml, Jackson Immunoreseach), and developed with a VECTOR Blue Alkaline Phosphatase Substrate Kit III (Vector Laboratories). Spots in each well were counted using the CTL immunospot reader (Cellular Technologies Ltd). Quantitative PCR for expression of select transcription factor genes from mRNA. Five thousand cells were sorted from each population as described above. Total cellular RNA was isolated using the RNeasy Mini Kit (Qiagen) by following the manufacturer’s protocol. Approximately 400 pg of RNA was reverse transcribed using the iScript RT Kit (Bio-Rad). Aliquots of the resulting single-stranded cDNA products were included with BLIMP-1, Pax5, or GAPDH TaqMan assays (assay IDs: Hs00153357_m1, Hs00172003_m1, Hs00231936_m1, and Hs02758991_g1; Life Technologies) using IQ Supermix (Bio-Rad) and amplified using a Bio-Rad CFX96 Real-time PCR Detection System for 50 cycles. Resulting Ct values were normalized to GAPDH levels and relative target mRNA for each population was calculated based on a standard curve created using total RNA from all populations. RNA transcriptome analysis. RNA was isolated from sorted ASC subsets from peripheral blood samples for 6 adult donors. RNA-seq was performed as recommended by the manufacturer (Illumina IIx). Briefly, first-hand cDNA was amplified using random hexamers. Following end repair, addition of adaptor ligation, agarose gel isolation of 200-bp cDNA, and PCR amplification of the 200-bp cDNA, the samples were sequenced on an Illumina HiSeq 2000. Over 25 million single-end sequences were obtained per sample, and aligned to the reference annotated human genome hg38 using STAR (54). Exon-level data for approximately 25,000 known gene sequences per sample were identified and consolidated using HTSeq (55) to yield gene-level expression values, then normalized to total counts per gene per million total aligned reads and subsequently TMM values in edgeR (56) that were converted to the log2 scale. The RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus database (GSE11697). Differential expression was assessed with the GLM ANOVA routine in JMP Genomics v8.0 (SAS Institute) after further normalization using the SNM procedure in R to remove batch effects with ASC population as the Biological variable and Batch as the adjustment variable with the Rm = True option (57). A lower threshold of 3 log2 units for inclusion was selected by plotting the coefficient of variance against average abundance. All analyses were qualitatively confirmed for the raw TMM values without SNM. Gene expression was visualized by hierarchical clustering using standardization of each gene across individuals and Ward’s method to weight the correlations in JMP Genomics. Pathway analysis. GSEA was performed using the Broad Institute’s preranked hallmark pathway gene lists (58, 59). For each of 29 significantly enriched pathways (FDR < 0.05; Kolgomorov-Smirnov nominal P < 0.05), the first principal component of the genes computed in R was used to visualize the overall regulation of the gene set. Because PC1 is somewhat arbitrarily signed, we compared these values with the direction of normalized enrichment and adjusted the sign to be concordant with an abundance of upregulation, contrasting pop 5 with pops 2 and 3. Because PC1 is dominated by strongly coregulated genes, it does not necessarily give the same results or significance values as normalized enrichment based on all of the genes in the pathway. An additional 6 pathways were excluded from further consideration because PC1 explained less than 20% of the variance, indicating little covariance; the 29 highlighted pathways all have PC1 explaining greater than 45% of the variance of the genes. Supplemental Figure 5 provides details for 12 gene sets (Allograft Rejection, Coagulation, Complement, DNA Repair, Heme Metabolism, IFNG Response, Mitotic Spindle, MTORC1 Signaling, Peroxisome, Spermatogenesis, UV Response Up, and Xenobiotic Metabolism) where there is some discordance between the two modes of analysis. Most cases are because PC1 is computed from both up- and downregulated genes that strongly differ among pops, whereas the enrichment score indicates the significance
Somatic hypermutation is important for the generation of high-affinity antibodies,
but this mutational process is also likely to negatively impact the functional integrity
of B cell receptors (BCRs). Stewart et al. find that germinal center B cells replace
surface BCRs in dark zones (DZ) and present evidence for a DZ checkpoint that prevents
the accumulation of clones with non-functional BCRs, thus facilitating selection in
the LZ.
Successful vaccination relies on driving the immune response towards high specificity, affinity and longevity. Germinal centers facilitate the evoluti…
Review Free access | 10.1172/JCI90962 The role of the complement system in cancer Vahid Afshar-Kharghan First published March 1, 2017 - More info Abstract In addition to being a component of innate immunity and an ancient defense mechanism against invading pathogens, complement activation also participates in the adaptive immune response, inflammation, hemostasis, embryogenesis, and organ repair and development. Activation of the complement system via classical, lectin, or alternative pathways generates anaphylatoxins (C3a and C5a) and membrane attack complex (C5b-9) and opsonizes targeted cells. Complement activation end products and their receptors mediate cell-cell interactions that regulate several biological functions in the extravascular tissue. Signaling of anaphylatoxin receptors or assembly of membrane attack complex promotes cell dedifferentiation, proliferation, and migration in addition to reducing apoptosis. As a result, complement activation in the tumor microenvironment enhances tumor growth and increases metastasis. In this Review, I discuss immune and nonimmune functions of complement proteins and the tumor-promoting effect of complement activation. Introduction The complement system is a cascade of serine proteases encoded by genes originating from the same ancestral genes as coagulation proteins (1). Like the coagulation system, complement activation involves several steps, is tightly regulated, and requires both plasma and membrane proteins (2, 3). Many complement proteins possess dual functions that provide crosstalk between the complement system and other effector and regulatory systems. As a result, the complement system participates in adaptive immunity, hemostasis, neuroprotection and synaptic pruning, and organ development in addition to its role in innate immunity. It is also involved in a diverse array of pathologic conditions, such as thrombotic disorders, autoimmune disorders, schizophrenia, alloimmune responses including allograft rejection and graft-versus-host disease, and cancer. The complement system’s role in fighting invasive pathogens has been extensively studied (4, 5), but recent discoveries provide new perspectives on the complement system’s function in the extravascular and interstitial tissue compartment. These discoveries illustrate an important role for complement proteins in cell-cell and stroma-cell communications. In this Review, I briefly discuss activation, regulation, immune, and nonimmune functions of the complement system to provide a framework for examining the role of complement in cancer. Activation of the complement system The complement system is activated by three major pathways: the classical pathway, via antigen-antibody complexes; the alternative pathway, via any permissive surfaces; and the lectin pathway, via binding of pattern-recognizing mannose-binding lectins (MBLs) to carbohydrate ligands on the surface of pathogens (Figure 1 and refs. 6–9). The convergence point for all complement activation pathways is the formation of the C3 convertase complex on the surface of targeted cells, summarized in Figure 1, A–C. After forming C3 convertase, complement is able to carry out its effector functions. Figure 1 Complement activation. (A) The classical pathway is initiated by a complement-fixing antibody binding to an antigen on targeted cells. C1q binds to the antibody’s Fc domain in the antibody-antigen complex. C1r and C1s assemble on C1q, C1r cleaves and activates C1s, and activated C1s cleaves C4 and C2 into C4b and C2a, respectively. C4b and C2a form the C3 convertase C4bC2a. (B) In the lectin pathway, MBL binds to repetitive sugar moieties such as mannose. MBL and MASP2 then form a C1-like complex. Activated MASP2 in MBL-MASP2 complex cleaves C4 and C2 and generates C3 convertase (C4bC2a). (C) In the alternative pathway, small amounts of hydrolyzed plasma C3 [C3(H2O)] bind to factor B, which forms the C3(H2O)Bb complex with help from factor D. C3(H2O)Bb cleaves additional plasma C3 to generate highly active C3b, which binds to cell the surface. On a complement-activating surface, C3b binds Bb (produced by factor D–mediated cleavage of factor B) and generates C3bBb (the alternative pathway’s C3 convertase). (D) Regardless of the initiation steps, C3 convertase deposits additional C3b molecules and generates C3a. If it remains intact, C3 convertase binds to additional C3b to generate C5 convertase. C5 convertase cleaves C5 to generate C5b. (E) C5b binds to C6, C7, and C8, forming a C5b-8 complex, which polymerizes several C9 molecules, forming the cytolytic MAC. In all three complement activation pathways, C3 convertase complex cleaves C3 molecules to C3a, one of the two major anaphylatoxins, and to C3b, a potent opsonin. Binding of C3b molecules to the surface of cells or cell debris in a process called opsonization marks them for phagocytosis by macrophages. Surface-bound C3b and its degradation products are ligands for complement receptors CR1, CR3, and CR2 that are expressed on myelomonocytic cells, lymphocytes, and follicular dendritic cells. Binding of C3b and its degradation products to correspondent receptors are crucial to cell-cell interactions in the innate and adaptive immune responses and in the removal of complement-coated apoptotic and necrotic cells. Propagation of complement activation by C3 convertase results in the generation of the C5 convertase complex on the cell surface. C5 convertase then cleaves C5 to C5a and C5b. C5a is a potent anaphylatoxin and recruits neutrophils to areas of inflammation and tissue damage. C5b forms a complex with C6 and C7 that may insert into cell membrane, and subsequently be joined by C8 and multiple C9 to form the membrane attack complex (MAC or C5b-9 complex; Figure 1D). Deposition of an adequate number of MACs disrupts the phospholipid bilayer of the cell membrane, leading to massive calcium influx, loss of mitochondrial membrane potential, and cell lysis. However, MAC deposition at sublytic concentrations on cell membrane has a different result, activating intracellular signal transduction and cell proliferation (10). Eukaryotic cells have developed several defense mechanisms to counteract the dire consequences of MAC accumulation at the cell surface, including expression of complement regulatory proteins (CRPs) that disassemble MAC (i.e., CD59, vitronectin, and clusterin), and endocytosis or shedding of MAC from the cell surface. Thus, the three main consequences of complement activation are tagging of cells by C3b degradation products for phagocytosis; chemotaxis of inflammatory cells in response to C3a and C5a; and MAC-mediated cell lysis. As described below, complement activation end products affect tumor growth by altering cancer cell behavior and modulating the immune response to the tumor. Regulation of the complement system The complement system’s ability to cause cellular damage is strictly controlled at several steps, both in the fluid phase and on the cell surface (6). In the classical and lectin pathways, C1 inhibitor (C1INH) binds to and inactivates C1r, C1s, and MBL-associated serine proteases (MASPs). The activities of other CRPs can be categorized into two major groups: (a) decay-accelerating activity, which breaks up the C3 convertase complex, as can be seen in C4-binding protein (C4bp), CR1, decay-accelerating factor (DAF, also known as CD55), and factor H; and (b) membrane cofactor activity, which acts as a cofactor for the factor I–mediated cleavage of C3b or C4b to their inactive degradation products, iC3b and iC4b, respectively. CRPs with membrane cofactor activity include C4bp, CR1, membrane cofactor protein (MCP, or CD46), and factor H. Another important CRP is CD59, which is expressed on many different cell types and prevents assembly of MAC on the cell membrane. The anaphylatoxins C3a and C5a are complement activation products that are rapidly inactivated in plasma by carboxypeptidases, particularly carboxypeptidase N (11). CRPs are overexpressed by many cancer cells and may be used as potential therapeutic targets. Immune function of the complement system The complement system is an ancient defense mechanism preceding adaptive immunity (12). It can be activated by pattern-recognition molecules and natural antibodies (13). Complement system activation and generation of anaphylatoxins orchestrate an inflammatory response to pathogens (12, 14). Anaphylatoxins activate macrophages, neutrophils, mast cells, basophils, and eosinophils, resulting in their degranulation and the production of cytokines, which in turn causes vasodilation, increases vascular permeability, and enhances neutrophil extravasation and chemotaxis (13). The complement system links innate immunity to adaptive immunity. Complement deficiency impairs both B and T cell responses (15). The effect of complement on the B cell response is mediated by CR2 on B cells and follicular dendritic cells. Activation of the classical pathway on the surface of an antigen tags that antigen with C3d, enabling its binding to CR2 on B cells. CR2, CD19, and CD81 form a B cell coreceptor complex, and CR2 engagement with C3d enhances signaling through antigen-encountered B cell receptors and decreases the activation threshold of B cells (12, 15). The interaction between CR2 on follicular dendritic cells and C3d on antigens is important for antigen presentation to naive and primed B cells in the germinal center of lymph nodes, in the maturation of B cells, and in the generation of memory B cells. The role of complement proteins in the cognate interaction between antigen-presenting cells (APCs) and T cells is important in the T cell immune response (16). In addition to systemic production in the liver, complement proteins are also produced locally by T cells and APCs (17–20). The effects of complement proteins on activation, proliferation, and differentiation of T cells are mediated by the local complement activation, by production of C3a and C5a at the interface of T cells and APCs, and through anaphylatoxin receptors on T cells and APCs (17–19, 21, 22). Reducing the number of C3a and C5a receptors (C3aR and C5aR, respectively) on T cells or APCs impairs T cell immunity. Complement proteins and receptors are involved in different stages of the interaction between APCs and T cells. APCs produce C3 and express C3aR and C5aR, both of which are essential for their maturation and differentiation (19) and for effective antigen presentation to T cells (17, 23, 24). C3- or C3aR-deficient APCs are much less potent in inducing a T cell immune response compared with WT APCs (19, 25). After APCs present antigen to T cells, C5aR on the T cells is required for their proliferation. Binding of C5a to C5aR on T cells has both antiapoptotic and pro-proliferative effects (22). Nonimmune function of the complement system Cell-cell and stroma-cell interactions mediated by complement proteins regulate several physiologic processes, such as collective cell migration during embryogenesis (26), synaptic pruning during brain development (27–30), cell proliferation and differentiation during liver regeneration (31) and bone development (32, 33), and hematopoietic stem cell migration and engraftment during hematopoiesis (34). Complement and cancer The surge of interest in cancer immunotherapy is mainly focused on manipulating function or number of cytotoxic T cells. However, two important reasons justify studying the role of complement activation in cancer progression and the effect of complement manipulation in cancer therapy. First, the complement system is an important component of the inflammatory response, and inflammation is involved in various stages of tumorigenesis and cancer progression (35). Second, complement activation regulates adaptive immune response (15) and might have a role in regulating T cell response to tumors. Complement system in inflammation and tumorigenesis. Tumor-promoting inflammation has an important role in carcinogenesis and cancer progression (36–38). A series of elegant experiments established that activation of the complement system is an important component of tumor-promoting inflammation. Bonavita et al. showed that C3-deficient mice were protected against chemical carcinogenesis in mesenchymal and epithelial tissues (39), mainly because of reduced inflammation. Authors identified a humoral component of innate immunity, the long pentraxin PTX3, as an important negative regulator of inflammation and complement activation. PTX3-deficient mice were susceptible to chemical carcinogenesis, displaying an increased number of tumor-associated macrophages with M2 phenotype and increased concentration of CCL2 chemokine inside tumors. The tumor-promoting inflammation induced by PTX3 deficiency was complement-dependent and completely reversed after removal of C3, as manifested by a reduction in the susceptibility of Ptx3–/– C3–/– mice to chemical carcinogenesis. Similarly, treatment with the C5aR antagonist PMX-53 reversed the susceptible phenotype of Ptx3–/– mice without affecting the rate of tumorigenesis in Ptx3+/+ mice. Activation and regulation of complement pathways in tumors. Expression of complement and CRPs is increased in malignant tumors and cancer cell lines (summarized in Table 1). Complement proteins, C3 degradation products, and complement activation products (i.e., C5a, C3a, and C5b-9) are easily detectable in various types of cancer, consistent with complement activation inside these tumors. Table 1 Complement proteins in cancer The main pathway involved in activation of complement inside tumors is unclear, and evidence supports activation of each complement pathway in malignant tumors (40). To make matters more complicated, cancer cell membrane-bound serine proteases can also cleave C5 and generate C5a without complement activation (41). Additionally, complement proteins expressed in tumors might also play a role in cancer progression independent of complement activation, as was shown for C1q in a syngeneic murine model of melanoma, where C1q expression affected angiogenesis, tumor progression, and metastasis (42). In this murine model, C1q was expressed in endothelial cells, spindle-shaped fibroblasts, and tumor-infiltrating myeloid cells independently of C4. Lack of C4 coexpression in C1q-expressing tumors hints at a role for C1q in tumor progression independent of the classical pathway. Expression of CRPs, including both membrane proteins (CD55, CD59, MCP, or CD46) and soluble proteins (factor H and factor H–like proteins), is increased in cancer cells (43), although the overexpression of CRPs is heterogeneous among different cancer types and even between different tumor specimens of the same type of cancer (44). One interpretation of the presence of both complement activation products and CRPs in tumors is that complement activation is a host defense mechanism against cancer, and cancer cells resist complement attack by overexpressing CRPs. However, as discussed later in this Review, several recent studies do not support this interpretation and suggest another scenario in which local complement activation inside tumors enhances tumor growth. Complement activation: antitumor or protumor? Evidence for the effects of complement on malignant transformation of epithelial cells and progression of cancer has evolved based on several recent studies showing complex and sometimes contradictory findings. This complexity is similar to the complex role of inflammation in cancer (45). Although inflammatory cells and cytokines are important in immune surveillance, exemplified by the benefit of bacillus Calmette-Guérin therapy in early stages of bladder cancer, chronic inflammation promotes carcinogenesis and tumor growth. Even immune cells, such as macrophages, can have both pro- and antitumor phenotypes. Despite this multifaceted picture, most evidence points toward a protumor effect of chronic inflammation (45). The long-held view of complement activation as an antitumor defense mechanism is based on two main concepts: first, the complement system’s participation in immune surveillance against malignant cells, and second, complement-dependent cytotoxicity of therapeutic monoclonal antibodies. I will discuss these concepts below, and summarize new information pointing toward a protumor effect of complement activation inside tumors. Complement and immune surveillance. The complement system’s ability to distinguish self from non-self makes it an important part of the innate immune response to invading pathogens (46). Expression of non-self antigens and lack of CRPs on microbes make them optimal targets for complement detection and, later on, complement-mediated elimination. Similarly, expression of danger signals and neoantigens by apoptotic cells and cellular debris optimizes their detection and removal by the complement system. Cancer cells, on the other hand, mostly express the same proteins as their normal epithelial cell counterparts, albeit occasionally with a different density. Furthermore, overexpression of CRPs by cancer cells limits immune surveillance by the complement system (3, 43, 46, 47). Putting these findings together, one can conclude that cell-mediated immunity plays a more important role than humoral immunity in immune surveillance against cancer cells (48, 49), and effectiveness of complement in early detection and elimination of cancer cells is uncertain (50). Complement-dependent cytotoxicity. Complement activation was considered detrimental to cancer cells via complement-dependent cytotoxicity, which causes cancer cell lysis via MAC accumulation or phagocytosis of opsonized cancer cells by macrophages and neutrophils. Complement-dependent cytotoxicity is considered to be the main mechanism for the effectiveness of antitumor monoclonal antibodies. Rituximab, an anti-CD20 antibody against malignant B cells, is among the oldest and most widely used therapeutic monoclonal antibodies. Although in vitro and in vivo studies show that rituximab activates the classical complement pathway (51, 52), the notion that its therapeutic benefits are mainly mediated by induction of complement attack on malignant B cells is questionable. In fact, the antitumor effect of rituximab was inhibited by deposited complement proteins on B cells (53), and was enhanced in complement-deficient mice (54). Therefore, the extent to which complement-dependent cytotoxicity contributes to other immunologic effects of rituximab, i.e., antibody-dependent cellular cytotoxicity and antibody-dependent phagocytosis, is unknown. Other studies on the therapeutic mechanism of rituximab also showed a complement-independent, proapoptotic effect mediated by cross-linking of CD20 (55), as well as antiproliferative and antisurvival effects that were mediated by inhibition of B cell receptors (56). Furthermore, many in vitro antitumor effects of complement-fixing antibodies on cancer cell lines were not reproduced in vivo (57). Complement activation promotes tumor growth. Considering that complement is not efficient in immune surveillance against cancer cells and that the main antitumor effect of monoclonal antibodies might not arise from complement activation, the data supporting an antitumor role for complement activation are scant. The question remains: If complement does not attack cancer cells, how does local complement activation and deposition of complement proteins affect tumors? To understand the consequence of complement activation inside tumors, it is helpful to reexamine the biological functions of complement activation products. C3b and its degradation products binding to CR1, CR2, and CR3 provide ligands and receptors for cell-cell and stroma-cell interactions in many physiologic and pathologic conditions. Complement activation generates C3a and C5a and MAC. The anaphylatoxin receptors C3aR and C5aR are G protein–coupled receptors present on many cell types, including lymphocytes, monocytes/macrophages, myeloid cells, hematopoietic stem cells, mesenchymal cells, and epithelial cells, including cancer cells. Anaphylatoxin receptor signaling has been studied extensively (58). Activation of C5aR promotes a range of responses depending on the cell type. Relevant to its role in cancer, C5aR activation generates prosurvival and antiapoptotic responses. For example, C5a binding to C5aR decreases apoptosis in neutrophils (59) and T cells (22), and increases cell proliferation in endothelial (60) and colon cancer cell lines (61). Activation of C3aR plays an important role in guiding collective cell migration (26) and epithelial-mesenchymal transition (62, 63), both important mechanisms in metastasis. In a sublytic density, MAC accumulation on the cell membrane promotes cell proliferation (64) and differentiation, inhibits apoptosis (10, 65), and protects cells against complement-mediated lysis (66). Markiewski et al. showed that the activation of the classical complement pathway inside implanted orthotopic tumors in mice enhanced tumor growth (67). Complement’s progrowth effect on tumors was C5a-dependent and was eliminated in C5aR-deficient mice and in WT mice treated with a C5aR antagonist. C5a modulates the immune response to tumors by acting as a chemotactic factor, increasing infiltration of myeloid-derived suppressor cells (MDSCs) and reducing the number of CD8+ cytotoxic T cells inside tumors. MDSCs are immature myeloid cells that increase in blood, bone marrow, and spleen of tumor-bearing mice and cancer patients (68, 69) and assist tumor cells in evading the antitumor immune response. MDSCs reduce proliferation and increase apoptosis in CD8+ T cells by generating ROS and reactive nitrogen species (70). Depletion of CD8+ T cells in mice eliminated the protective effect of complement deficiency against tumor growth. In summary, this study showed that the immunomodulatory effect of activated classical complement pathway inside tumors enhances tumor growth. The origin of complement proteins was the host, but activation of complement occurred inside the tumor microenvironment, and the final effect on the tumor was an indirect immunomodulatory effect mediated by MDSCs (Figure 2). Figure 2 Effect of complement activation in the tumor microenvironment. Activation of the complement system inside tumors releases C5a and C3a into the tumor microenvironment and promotes tumor growth. C5a attracts myeloid cell, including MDSCs, into the tumor. MDSCs then reduce cytotoxic T cell responses to the tumor by inducing apoptosis and inhibiting CD8+ TILs via generation of ROS and reactive nitrogen species and depletion of arginine. In melanoma, secretion of C3 by CD8+ TILs and complement activation in the vicinity of these cells reduce IL-10 production by TILs and inhibit their function. Some cancer cell types secrete complement proteins into the tumor microenvironment and initiate an autocrine loop that increases cell proliferation and promotes metastasis. The effect of complement activation on MDSCs, TILs, and cancer cells is mediated by the C5a and C3a receptors (C5aR and C3aR) on these cells. In a follow-up study, Nunez-Cruz et al. investigated complement’s role in tumorigenesis in a murine model of spontaneous ovarian cancer (71, 72). C3 or C5aR deficiency in these mice prevented the development of ovarian tumors, permitting no tumors or only small and poorly vascularized tumor formation (71). C3 deficiency was associated with a change in the immune profile of leukocytes infiltrating into the tumors, but C5aR deficiency reduced ovarian tumor size without altering the immune profile of infiltrating leukocytes. This result suggested the existence of a protumor effect of complement that was independent of its immunomodulatory effect. We investigated the effect of complement in murine models of ovarian cancer and confirmed activation of complement in the tumor microenvironment (73). However, complement proteins detected inside ovarian tumors originated not from the host, but from tumor cells themselves. Complement activation products were present even inside tumors implanted in C3-deficient mice lacking a functional complement system. Although orthotopic ovarian cancer tumors in C3-deficient mice reached to the same size as those in WT mice, reducing C3 or C5 production in cancer cells significantly reduced the tumor growth independent of the host’s complement sufficiency status. C3 synthesis can be detected in malignant epithelial cells originating from several different organs, particularly lung and ovary. Inhibiting synthesis of complement proteins in cancer cells altered the immune profile of leukocytes infiltrating into tumors, manifested by an increase in the number of CD8+ T cells and reduction in myeloid cells. However, immunomodulatory effect of complement inhibition was not the main mechanism responsible for the observed reduction in tumor growth. Inhibiting complement protein synthesis in cancer cells implanted in CD8+ T cell–deficient mice reduced tumor growth to the same magnitude as in WT mice. We investigated the possibility of an autocrine stimulation of cancer cells as a result of complement activation. Anaphylatoxin receptors are present on ovarian cancer cells, and stimulation of these receptors by C3a or C5a agonist peptides increased proliferation and invasiveness of ovarian cancer cells in vitro. Furthermore, knockdown of these receptors on cancer cells reduced growth of orthotopic ovarian tumors in mice. Our studies showed that local complement activation inside the tumor microenvironment enhances tumor growth via a direct autocrine effect on ovarian cancer cells increasing cell proliferation (Figure 2). In a murine model, Wang et al. reported another mechanism for the progrowth effect of complement activation in melanoma, showing that production of IL-10 by CD8+ tumor-infiltrating lymphocytes (TILs) is constitutively inhibited in an autocrine fashion by C3 originating from CD8+ TILs themselves, acting through C5aR and C3aR on the surface of these lymphocytes (74). C3aR and C5aR antagonists increased IL-10 production and activated CD8+ TILs that in turn reduce tumor growth. The IL-10–dependent antitumor activity of complement inhibitors in melanoma was independent of the PD-1/PD-L1 axis or MDSCs. This study provides evidence that local complement activation in the tumor microenvironment results in suppression of the immune response to melanoma by inhibiting CD8+ TIL function (Figure 2). The studies above describe different mechanisms by which complement activation in the tumor microenvironment can enhance tumor growth: (a) by altering the immune profile of tumor-infiltrating leukocytes, (b) by increasing cancer cell proliferation, and (c) by directly suppressing CD8+ TIL function. It is possible that different cancer types use different mechanisms to take advantage of ectopic complement activation inside tumors. For example, ovarian cancer cells synthesize a significant amount of complement proteins and initiate an autocrine loop resulting in increased cell proliferation by a direct effect of anaphylatoxins on cancer cells. Conversely, melanoma cells do not secrete complement proteins, and complement proteins produced by CD8+ TILs reduce their IL-10 production and antitumor activity. An important question remains whether complement activation has any role in malignant transformation of normal cells or only affects the expansion of already established malignant clones. Most available data are based on orthotopic murine models of ovarian cancer or mice genetically engineered to develop ovarian cancer by overexpression of oncogenes. These studies showed that complement promotes growth and expansion of malignant tumors. Bonavita et al. showed that complement promotes malignant transformation of cells exposed to chronic inflammation induced by chemical carcinogens (39). However, additional studies are required to dissect the effect of early versus late stages of complement activation on various stages of oncogenesis. Overexpression of CRPs on cancer cells If complement activation promotes tumor growth and oncogenesis, why are CRPs overexpressed on cancer cells? One would expect that cancer cells, under selective pressure, downregulate expression of CRPs to benefit from complement activation. To reconcile these seemingly counterintuitive observations, we put forward the following hypothesis: Anaphylatoxins and sublytic concentrations of MAC promote tumor growth, but higher concentrations of MAC have a tumoricidal effect. As a result, cancer cells benefit from early stages of complement activation and production of anaphylatoxins, but actively inhibit generation of MAC. Cancer cells reduce MAC concentration by overexpressing CD59, the most consistently overexpressed CRP on different cancer cells (75) and the most effective membrane regulatory protein against complement-mediated lysis (43, 76, 77), eliminating MAC from the cell surface through membrane vesiculation. Thus, from a therapeutic point of view, interventions that reduce complement activation or promote the generation of MAC inside tumors can be considered as logical options to counter the progrowth effect of complement on cancer. Complement activation in epithelial-mesenchymal transition Epithelial-mesenchymal transition (EMT) occurs in physiologic processes such as embryogenesis and organ development, and in pathologic conditions including tissue fibrosis and metastasis (78, 79). Complement participates in EMT in murine models of renal injury and fibrosis (62, 80, 81). We showed that complement activation inside tumors not only increases tumor growth but also enhances metastasis by promoting EMT in cancer cells. In ovarian cancer cells, the transcription factor TWIST1 upregulates C3 gene expression, generating C3a in the tumor microenvironment, which binds to C3aR on ovarian cancer cells. We further showed that C3aR signaling increases EMT and decreases E-cadherin expression in ovarian cancer cells via a Krüppel-like factor 5–dependent mechanism and promotes EMT and metastasis (63). In addition to promoting metastasis, EMT also induces resistance to complement-dependent cytotoxicity in lung cancer cells by increasing expression of CD59 (82). Inhibition or knockdown of CD59 restored sensitivity of cancer cells to complement-dependent lysis without altering the morphologic features or protein markers of EMT in these cells. Therapeutic potential of targeting complement activation in cancer Our understanding of the role of the complement system in cancer biology is evolving, changing our approach to the therapeutic use of reagents modifying the complement system. Traditional methods targeting cancer cells using antitumor antibodies to promote lysis of cancer cells by MACs require identification of tumor-specific antigens that either are expressed with a higher density on cancer cells than normal epithelial cells, or are only expressed on cancer cells. Antibodies against EGFR and CD20 are among the most successful therapeutic antibodies. Development of therapeutic antibodies was initially complicated by induction of an immune response to polyclonal antibodies developed in nonhuman hosts. Development of monoclonal murine antibodies; later, chimeric human-mouse antibodies; and recently, humanized antibodies (83) helps overcome this problem. However, a more important problem in harvesting complement-dependent cytotoxicity induced by therapeutic antibodies is overexpression of membrane CRPs by cancer cells that let cancer cells evade MAC-mediated cytolysis (43). As a result, blockade of membrane CRPs on cancer cells, alone or in conjunction with use of therapeutic antibodies, has been tried as another potential therapeutic strategy. Blocking CRPs reduces cancer cell proliferation in vitro (43) and tumor growth in mice (76). CD59 blocking antibodies or CD59 siRNA enhanced complement-mediated cytolysis induced by anti-EGFR monoclonal antibodies (trastuzumab and cetuximab) in human lung cancer cell lines (84). rILYd4, a recombinant protein inhibitor of CD59, increased sensitivity of malignant B cells to rituximab in vitro and in orthotopic murine models (85). Membrane CRPs are universally expressed, and an important theoretical complication of blocking CRPs is exposing normal cells to complement-dependent cytotoxicity. For example, CD59 and CD55 protect red blood cells against complement-induced hemolysis, and blocking CD59 might cause hemolysis. Interestingly, administration of rILYd4 in mice was not associated with significant increases in hemolysis (85). More recent studies showed a protumor effect of complement, and inhibition of complement activation in vitro or in murine models of cancer was investigated as a novel way to treat cancer. Understanding of the autocrine and paracrine effects of complement production and activation inside tumors versus its systemic immunomodulatory effect is not complete, but blocking complement activation or inhibiting C5aR and C3aR signaling inside tumors seems a reasonable approach. However, several questions and concerns regarding the therapeutic use of anticomplement reagents have not been addressed and require additional studies: Does systemic complement inhibition affect local production and activation of complement in the tumor microenvironment? Pharmacokinetic studies based on measurements of tissue concentration of various anticomplement reagents may resolve this issue, although the leakiness of tumor vasculature likely provides adequate tissue penetrance of these reagents. Does complement have a protumor effect in many or only in a few types of cancer? Recent studies showed that complement activation enhances growth of ovarian, cervical, and non–small-cell lung cancer and sarcoma, and deposition of complement proteins is detectable in more cancer cell types (Table 1). Which complement pathways are activated in cancer? Evidence supporting activation of classical, lectin, and alternative pathways in cancer exists. It is possible that in different types of cancer different pathways are functional. Therefore, it is more reasonable to target common complement proteins or receptors in antitumor therapies. What are the effects of early (C3a and C5a) versus late (MAC) complement activation end products on cancer cells? If higher concentrations of C3a or C5a promote, and denser MAC deposit reduces, tumor growth, developing bispecific inhibitory antibodies targeting C5aR (or C3aR) and CD59 simultaneously may increase the potency of the antitumor effect of complement therapy. Complement-dependent cytotoxicity has been considered an important component of the therapeutic benefit of monoclonal antibodies in malignant B cell disorders; however, the effects of complement activation on white blood cell dyscrasia have not been studied. A few reports point to a prognostic significance of expression of complement genes in leukemic blasts (86, 87). More comprehensive studies on the role of complement activation in leukemia and lymphoproliferative disorders might reveal possible therapeutic benefits of anticomplement reagents in these disorders. Eculizumab, a humanized anti-C5 monoclonal antibody, is currently available on the market and is used to treat paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. A single i.v. infusion of eculizumab blocks complement activation in plasma for 2–3 weeks (88, 89), but its potency and half-life in the interstitial tissue are unknown. Eculizumab blocks generation of C5a and MAC, but would not affect synthesis and secretion of complement proteins by cancer cells or C3a generation in the tumor microenvironment. Currently, no ongoing clinical trials are evaluating eculizumab in cancer patients; however, because of the clinical use of this reagent for other indications, we have a relatively clear picture of its side effect profile. Patients on eculizumab are at risk for developing infections with encapsulated microorganisms and should receive meningococcal vaccination before initiation of therapy. Lack of bone marrow suppression with eculizumab is a therapeutic advantage that can be used in designing clinical trials combining this reagent with chemotherapeutic reagents in cancer patients. In a few animal studies, C5aR antagonists, including PMX-53, have been shown to be effective in reducing tumor size in mice (67, 71), but this or similar reagents have not entered into clinical practice yet. Targeting C5aR rather than C5 or C3 might have the potential benefit of leaving opsonization and MAC generation intact. Intact opsonization of bacteria would reduce the risk of infectious complications in individuals undergoing treatment, and the generation of lytic concentrations of MAC might have a tumoricidal effect. On the other hand, targeting C5aR has the disadvantage of leaving other complement effector molecules, such as C3a, uninhibited. A potential advantage of using anticomplement reagents in cancer treatment is that they can be combined with traditional chemotherapies without increasing myelosuppression associated with chemotherapies; and combined with immune checkpoint inhibitors, because they have different targets. While checkpoint inhibitors increase proliferation of cytotoxic T cells, complement inhibitors decrease MDSCs infiltrating into the tumor microenvironment, reduce MDSC-induced T cell suppression, and enhance T cell function. Based on experiences collected with the clinical use of eculizumab, another advantage of complement inhibitors is their relatively few side effects. Any therapeutic use of anticomplement therapies in solid or liquid tumors should be carefully balanced with possible interference of complement inhibition with the efficacy of other antitumor reagents: (a) The outcome of combining anticomplement reagents with monoclonal antibodies (such as cetuximab, rituximab, or trastuzumab) may depend on the importance of complement-dependent cytotoxicity in the function of these antibodies. (b) Chimeric antigen receptor (CAR) T cell therapies depend on in vitro expansion and in vivo proliferation of T cells. Complement inhibition may decrease the proliferation of CAR T cells in vivo and may reduce their efficacy. Conclusions By mediating cell-cell and cell-stroma interactions, complement proteins have several immune and nonimmune functions in both plasma and the extravascular interstitial tissue. Activation of the complement system in the tumor microenvironment enhances tumor growth via different mechanisms. Anticomplement reagents might have a place in the therapeutic armamentarium against cancer and, because of their limited non-myelosuppressive side effects and nonoverlapping pharmacodynamics, could be combined with traditional chemotherapies or immunotherapies. Acknowledgments This work is supported in part by NIH grant CA177909 (to VAK). The author thanks Michael Kroll for his valuable comments. Footnotes Conflict of interest: The author has declared that no conflict of interest exists. Reference information: J Clin Invest. 2017;127(3):780–789. https://doi.org/10.1172/JCI90962. References Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002;27(2):67–74. View this article via: PubMed CrossRef Google Scholar Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30(2):83–90. View this article via: PubMed CrossRef Google Scholar Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9(10):729–740. View this article via: PubMed Google Scholar Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–1066. View this article via: PubMed CrossRef Google Scholar Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–1144. View this article via: PubMed CrossRef Google Scholar Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol. 2014;32:433–459. View this article via: PubMed CrossRef Google Scholar Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40(7):423–429. View this article via: PubMed CrossRef Google Scholar Jack DL, Klein NJ, Turner MW. Mannose-binding lectin: targeting the microbial world for complement attack and opsonophagocytosis. Immunol Rev. 2001;180:86–99. View this article via: PubMed CrossRef Google Scholar Frank MM. Complement in the pathophysiology of human disease. N Engl J Med. 1987;316(24):1525–1530. View this article via: PubMed CrossRef Google Scholar Tegla CA, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. 2011;51(1):45–60. View this article via: PubMed CrossRef Google Scholar Matthews KW, Mueller-Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40(11):785–793. View this article via: PubMed CrossRef Google Scholar Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50. View this article via: PubMed CrossRef Google Scholar Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171(3):715–727. View this article via: PubMed CrossRef Google Scholar Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190(8):3831–3838. View this article via: PubMed CrossRef Google Scholar Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37(2):199–207. View this article via: PubMed CrossRef Google Scholar Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7(1):9–18. View this article via: PubMed CrossRef Google Scholar Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425–435. View this article via: PubMed CrossRef Google Scholar Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8(+) T-cell responses to allogeneic endothelial cells are controlled by local complement activation. Am J Transplant. 2009;9(8):1784–1795. View this article via: PubMed CrossRef Google Scholar Peng Q, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. 2008;111(4):2452–2461. View this article via: PubMed CrossRef Google Scholar Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582–587. View this article via: PubMed CrossRef Google Scholar Sacks SH. Complement fragments C3a and C5a: the salt and pepper of the immune response. Eur J Immunol. 2010;40(3):668–670. View this article via: PubMed CrossRef Google Scholar Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. View this article via: PubMed CrossRef Google Scholar Jacquier-Sarlin MR, Gabert FM, Villiers MB, Colomb MG. Modulation of antigen processing and presentation by covalently linked complement C3b fragment. Immunology. 1995;84(1):164–170. View this article via: PubMed Google Scholar Kerekes K, Cooper PD, Prechl J, Józsi M, Bajtay Z, Erdei A. Adjuvant effect of gamma-inulin is mediated by C3 fragments deposited on antigen-presenting cells. J Leukoc Biol. 2001;69(1):69–74. View this article via: PubMed Google Scholar Zhou W, Medof ME, Heeger PS, Sacks S. Graft-derived complement as a mediator of transplant injury. Curr Opin Immunol. 2007;19(5):569–576. View this article via: PubMed CrossRef Google Scholar Carmona-Fontaine C, et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell. 2011;21(6):1026–1037. View this article via: PubMed CrossRef Google Scholar Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. View this article via: PubMed CrossRef Google Scholar Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. View this article via: PubMed CrossRef Google Scholar Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16(12):1773–1782. View this article via: PubMed CrossRef Google Scholar Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. View this article via: PubMed CrossRef Google Scholar Strey CW, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198(6):913–923. View this article via: PubMed CrossRef Google Scholar Mastellos D, Lambris JD. Complement: more than a ‘guard’ against invading pathogens? Trends Immunol. 2002;23(10):485–491. View this article via: PubMed CrossRef Google Scholar Del Rio-Tsonis K, Tsonis PA, Zarkadis IK, Tsagas AG, Lambris JD. Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J Immunol. 1998;161(12):6819–6824. View this article via: PubMed Google Scholar Reca R, et al. Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing-related responses to SDF-1. Blood. 2003;101(10):3784–3793. View this article via: PubMed CrossRef Google Scholar Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. View this article via: PubMed CrossRef Google Scholar Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–291. View this article via: PubMed CrossRef Google Scholar Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. View this article via: PubMed CrossRef Google Scholar Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. View this article via: PubMed CrossRef Google Scholar Bonavita E, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160(4):700–714. View this article via: PubMed CrossRef Google Scholar Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. 2013;25(1):54–64. View this article via: PubMed CrossRef Google Scholar Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. 2014;32(4):1715–1719. View this article via: PubMed Google Scholar Bulla R, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. 2016;7:10346. View this article via: PubMed Google Scholar Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40(2–4):109–123. View this article via: PubMed Google Scholar Gancz D, Fishelson Z. Cancer resistance to complement-dependent cytotoxicity (CDC): Problem-oriented research and development. Mol Immunol. 2009;46(14):2794–2800. View this article via: PubMed CrossRef Google Scholar Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. View this article via: PubMed CrossRef Google Scholar Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. View this article via: PubMed CrossRef Google Scholar Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20(12):576–582. View this article via: PubMed CrossRef Google Scholar Nishioka K, Kawamura K, Hirayama T, Kawashima T, Shimada K. The complement system in tumor immunity: significance of elevated levels of complement in tumor bearing hosts. Ann N Y Acad Sci. 1976;276:303–315. View this article via: PubMed CrossRef Google Scholar Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22(1):23–32. View this article via: PubMed CrossRef Google Scholar Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009;30(6):286–292. View this article via: PubMed CrossRef Google Scholar Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. 2016;28(3):309–316. View this article via: PubMed CrossRef Google Scholar Di Gaetano N, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171(3):1581–1587. View this article via: PubMed CrossRef Google Scholar Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111(3):1456–1463. Wang SY, et al. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood. 2009;114(26):5322–5330. Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood. 2002;99(4):1314–1319. Kheirallah S, et al. Rituximab inhibits B-cell receptor signaling. Blood. 2010;115(5):985–994. Prang N, et al. Cellular and complement-dependent cytotoxicity of Ep-CAM-specific monoclonal antibody MT201 against breast cancer cell lines. Br J Cancer. 2005;92(2):342–349. View this article via: PubMed Google Scholar Ward PA. Functions of C5a receptors. J Mol Med (Berl). 2009;87(4):375–378. View this article via: PubMed CrossRef Google Scholar Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61(2):456–463. View this article via: PubMed CrossRef Google Scholar Kurihara R, et al. C5a promotes migration, proliferation, and vessel formation in endothelial cells. Inflamm Res. 2010;59(8):659–666. View this article via: PubMed CrossRef Google Scholar Cao Q, McIsaac SM, Stadnyk AW. Human colonic epithelial cells detect and respond to C5a via apically expressed C5aR through the ERK pathway. Am J Physiol Cell Physiol. 2012;302(12):C1731–C1740. View this article via: PubMed CrossRef Google Scholar Zhou X, et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol. 2013;305(7):F957–F967. View this article via: PubMed CrossRef Google Scholar Cho MS, et al. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J Immunol. 2016;196(3):1412–1418. View this article via: PubMed CrossRef Google Scholar Niculescu F, Badea T, Rus H. Sublytic C5b-9 induces proliferation of human aortic smooth muscle cells: role of mitogen activated protein kinase and phosphatidylinositol 3-kinase. Atherosclerosis. 1999;142(1):47–56. View this article via: PubMed CrossRef Google Scholar Soane L, Cho HJ, Niculescu F, Rus H, Shin ML. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001;167(4):2305–2311. View this article via: PubMed CrossRef Google Scholar Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. 2001;123(3):366–374. View this article via: PubMed CrossRef Google Scholar Markiewski MM, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–1235. View this article via: PubMed CrossRef Google Scholar Peranzoni E, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22(2):238–244. View this article via: PubMed CrossRef Google Scholar Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res. 2012;54(1-3):275–285. View this article via: PubMed CrossRef Google Scholar Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–999. View this article via: PubMed CrossRef Google Scholar Nunez-Cruz S, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14(11):994–1004. View this article via: PubMed CrossRef Google Scholar Connolly DC, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63(6):1389–1397. View this article via: PubMed Google Scholar Cho MS, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6(6):1085–1095. View this article via: PubMed CrossRef Google Scholar Wang Y, et al. et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. 2016;6(9):1022–1035. View this article via: PubMed CrossRef Google Scholar Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2017;222(1):45–54. View this article via: PubMed CrossRef Google Scholar Shi XX, Zhang B, Zang JL, Wang GY, Gao MH. CD59 silencing via retrovirus-mediated RNA interference enhanced complement-mediated cell damage in ovary cancer. Cell Mol Immunol. 2009;6(1):61–66. View this article via: PubMed CrossRef Google Scholar Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003;131(2):254–263. View this article via: PubMed CrossRef Google Scholar Lovisa S, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21(9):998–1009. View this article via: PubMed CrossRef Google Scholar Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. View this article via: JCI PubMed CrossRef Google Scholar Wan J, Zhou X, Cui J, Zou Z, Xu Y, You D. Role of complement 3 in TNF-α-induced mesenchymal transition of renal tubular epithelial cells in vitro. Mol Biotechnol. 2013;54(1):92–100. View this article via: PubMed CrossRef Google Scholar Zhou M, Ma H, Lin H, Qin J. Induction of epithelial-to-mesenchymal transition in proximal tubular epithelial cells on microfluidic devices. Biomaterials. 2014;35(5):1390–1401. View this article via: PubMed CrossRef Google Scholar Goswami MT, et al. Regulation of complement-dependent cytotoxicity by TGF-β-induced epithelial-mesenchymal transition. Oncogene. 2016;35(15):1888–1898. View this article via: PubMed CrossRef Google Scholar Macor P, Tedesco F. Complement as effector system in cancer immunotherapy. Immunol Lett. 2007;111(1):6–13. View this article via: PubMed CrossRef Google Scholar Zhao WP, Zhu B, Duan YZ, Chen ZT. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol Rep. 2009;21(6):1405–1411. Hu W, et al. Human CD59 inhibitor sensitizes rituximab-resistant lymphoma cells to complement-mediated cytolysis. Cancer Res. 2011;71(6):2298–2307. View this article via: PubMed CrossRef Google Scholar Laverdière I, et al. Complement cascade gene expression defines novel prognostic subgroups of acute myeloid leukemia. Exp Hematol. 2016;44(11):1039–1043.e10. View this article via: PubMed CrossRef Google Scholar Abdelbaset-Ismail A, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. [published online ahead of print August 26, 2016]. Leukemia. https://doi.org/10.1038/leu.2016.198. View this article via: PubMed CrossRef Google Scholar Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V, , French Study Group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643–657. View this article via: PubMed CrossRef Google Scholar Cugno M, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost. 2014;12(9):1440–1448. View this article via: PubMed CrossRef Google Scholar Canales NA, et al. A1BG and C3 are overexpressed in patients with cervical intraepithelial neoplasia III. Oncol Lett. 2014;8(2):939–947. View this article via: PubMed Google Scholar Lin K, et al. Complement component 3 is a prognostic factor of non–small cell lung cancer. Mol Med Rep. 2014;10(2):811–817. View this article via: PubMed Google Scholar Bouwens TA, Trouw LA, Veerhuis R, Dirven CM, Lamfers ML, Al-Khawaja H. Complement activation in Glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol. 2015;278:271–276. View this article via: PubMed CrossRef Google Scholar Nabizadeh JA, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. 2016;196(11):4783–4792. View this article via: PubMed CrossRef Google Scholar Habermann JK, et al. Increased serum levels of complement C3a anaphylatoxin indicate the presence of colorectal tumors. Gastroenterology. 2006;131(4):1020–1029; quiz 1284. View this article via: PubMed Google Scholar Bjørge L, et al. Ascitic complement system in ovarian cancer. Br J Cancer. 2005;92(5):895–905. View this article via: PubMed CrossRef Google Scholar Corrales L, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189(9):4674–4683. View this article via: PubMed CrossRef Google Scholar Nitta H, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin Cancer Res. 2013;19(8):2004–2013. View this article via: PubMed CrossRef Google Scholar Baatrup G, Qvist N, Junker A, Larsen KE, Zimmermann-Nielsen C. Activity and activation of the complement system in patients being operated on for cancer of the colon. Eur J Surg. 1994;160(9):503–510. View this article via: PubMed Google Scholar Ajona D, et al. Complement activation product C4d in oral and oropharyngeal squamous cell carcinoma. Oral Dis. 2015;21(7):899–904. View this article via: PubMed CrossRef Google Scholar Chen J, Yang WJ, Sun HJ, Yang X
Ise et al. identify the plasma cell-prone LZ GC B cells whose generation relies on
the amount of CD40 signal. Higher expression of ICAM-1 and SLAM in those cells facilitates
more stable contacts with Tfh cells, suggesting that strength of Tfh-GC B cell interaction
critically regulates formation of plasma cell precursors.
In this issue of JEM, Álvarez-Prado et al. (https://doi.org/10.1084/jem.20171738) designed a DNA capture library allowing them to identify 275 genes targeted by AID in mouse germinal center B cells. Using the molecular features of these genes to feed a machine-learning algorithm, they determined that high-density RNA PolII and Spt5 binding—found in 2.3% of the genes—are the best predictors of AID specificity.

|
Suggested by
LIGHTING
|
Luo et al. show that CD40 and BCR signaling in GC B cells is rewired to control very
different pathways, and both signals are required for optimal induction of c-Myc,
suggesting a mechanism of signaling-directed positive selection of GC B cells.
Although memory B cells sustain long-term humoral immunity, the nature of their precursors
within the germinal center has remained elusive. Suan et al. demonstrate that these
cells are uniquely identified by CCR6 expression in both mouse and human germinal
centers, that they are the most quiescent B cells in these structures, and that they
are generated within the light zone. Memory B cell precursors have a primarily low
affinity for antigen but also include cells emerging from the high-affinity compartment.
|
|
Suggested by
LIGHTING
|
Follicular helper T (Tfh) cells promote germinal center (GC) B cell survival and proliferation and guide their differentiation and immunoglobulin isotype switching by delivering contact-dependent and soluble factors, including IL-21, IL-4, IL-9, and IFN-γ. IL-21 and IFN-γ are coexpressed by Tfh cells during viral infections, but transcriptional regulation of these cytokines is not completely understood. In this study, we show that the T helper type 1 cell (Th1 cell) transcriptional regulators T-bet and STAT4 are coexpressed with Bcl6 in Tfh cells after acute viral infection, with a temporal decline in T-bet in the waning response. T-bet is important for Tfh cell production of IFN-γ, but not IL-21, and for a robust GC reaction. STAT4, phosphorylated in Tfh cells upon infection, is required for expression of T-bet and Bcl6 and for IFN-γ and IL-21. These data indicate that T-bet is expressed with Bcl6 in Tfh cells and is required alongside STAT4 to coordinate Tfh cell IL-21 and IFN-γ production and for promotion of the GC response after acute viral challenge.
The germinal center (GC) reaction begins with a diverse and expanded group of B cell clones bearing a wide range of antibody affinities. During GC colonization, B cells engage in long-lasting interactions with T follicular helper (Tfh) cells, a process that depends on antigen uptake and antigen presentation to the Tfh cells. How long-lasting T–B interactions and B cell clonal expansion are regulated by antigen presentation remains unclear. Here, we use in vivo B cell competition models and intravital imaging to examine the adhesive mechanisms governing B cell selection for GC colonization. We find that intercellular adhesion molecule 1 (ICAM-1) and ICAM-2 on B cells are essential for long-lasting cognate Tfh–B cell interactions and efficient selection of low-affinity B cell clones for proliferative clonal expansion. Thus, B cell ICAMs promote efficient antibody immune response by enhancement of T cell help to cognate B cells.
|